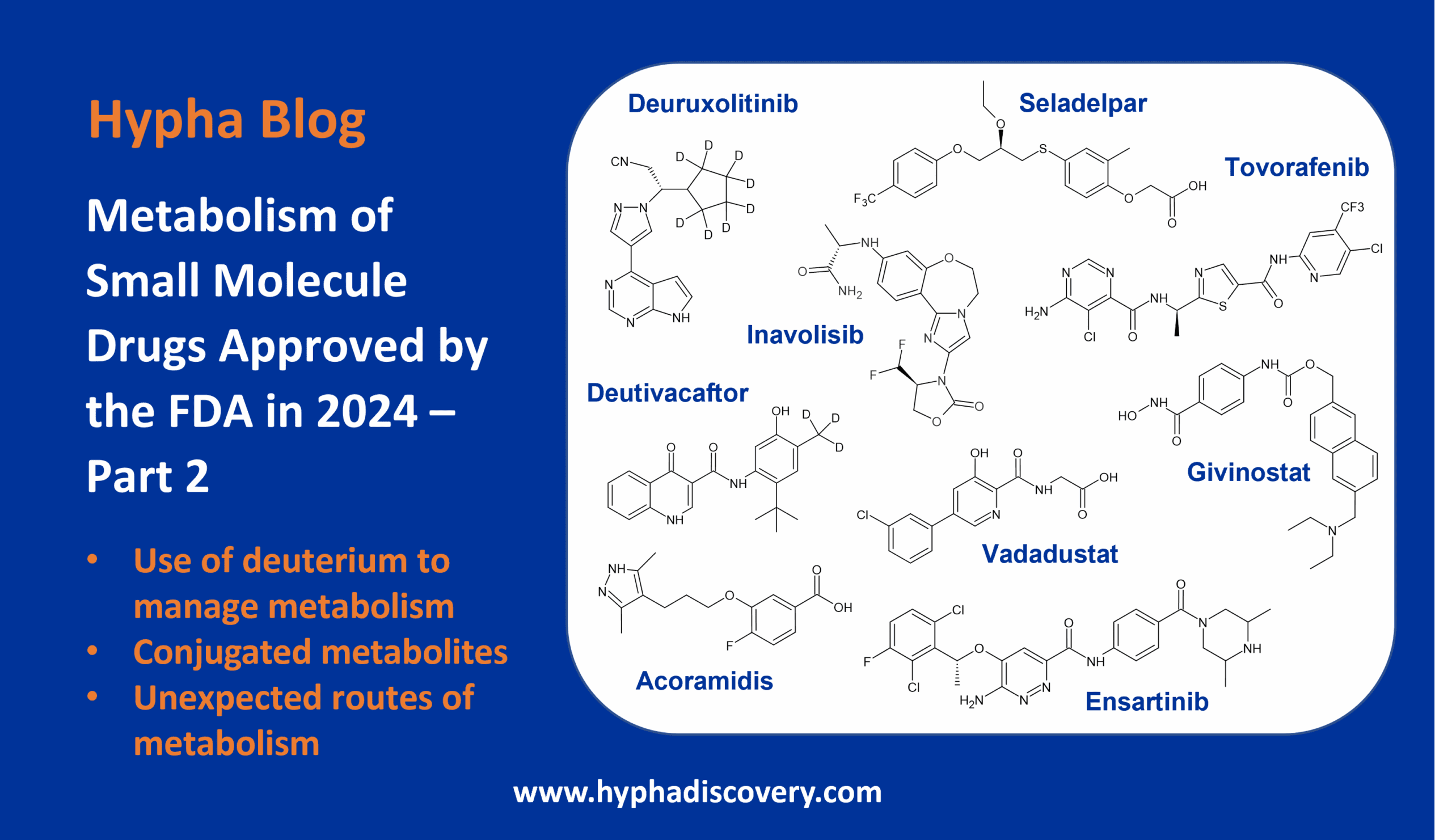
Metabolism of 2022 FDA approved small molecule drugs – Part 1
Does CYP3A4 still rule?
By Julia Shanu-Wilson
It won’t come as much surprise to learn that of the 17 small molecules* approved by the FDA in 2022, CYP3A4 was the major player in drug metabolism. Nonetheless, there are some interesting facets to discover, a selection of which we take a look at in a two-part blog on “Metabolism of 2022 FDA approved small molecule drugs.”
Analysis of the 100 most commonly prescribed drugs in 2022 revealed that, of the 89 subject to drug metabolism, 75% were cleared via phase I mechanisms, with CYP3A4 the major enzyme involved in metabolism of 48% of drugs (Iversen et al., 2022). This is pretty similar to what we observe for 2022 FDA new approved small molecules, but perhaps not surprising given we are looking at molecules that emerged from lab benches many years ago.
Table 1 – Small molecule drugs approved by the FDA in 2022 and the major reported route of biotransformation

*Note: 17 small molecule drugs as classified in C&EN (https://cen.acs.org/pharmaceuticals/37-new-drugs-achieved-FDA/101/i3 i.e. excluding antibody-drug conjugates, antibodies, proteins, radioligands and small interfering RNA).
What’s clear from the table is the lack of major metabolism via CYP2D6, despite this isoform being responsible for clearance of at least 20% of compounds in current clinical use (Maréchal et al., 2008). Since CYP2D6 displays a genetic polymorphism, resulting in large interindividual and ethnic differences, pharmaceutical companies have a motivation to design drug candidates that avoid metabolism by this CYP, something that seems to have come to fruition in this set of new drug approvals. Likely linked to this are the scaffolds taken through to clinical developed, since a feature common to the vast majority of CYP2D6 substrates is a basic nitrogen atom and a planar aromatic ring that need to fit in a rather buried active site, compared to the larger and more flexible active site of CYP3A4.
For the remainder of Part 1, we take a look at the metabolism of the drugs largely mediated by CYP3A4.
CYP3A4 mediated biotransformations
Daridorexant (ACT-541468) is one of several drugs approved whose clearance is mediated by CYP3A4. Similar to other dual orexin receptor antagonists suvorexant and almorexant, it is extensively metabolised by CYP3A4. An incredible 77 metabolites were detected across plasma, urine and faeces by AMS – microtracer / accelerator mass spectrometry (Muehlan et al., 2019), although many of these are very minor. The 3 main metabolites in human plasma have a much lower affinity to the OX1 and OX2 receptors compared to the parent drug, and do not appear to contribute to a significant extent to the pharmacological effect.

Figure 1: Biotransformation of daridorexant by CYP3A4
Major metabolites of daridorexant (≥10% of total drug-related exposure)
M1: aldehyde resulting from further oxidation of M3 at the benzylic position
M3: mono oxidised benzylic alcohol
M10: doubly oxidised metabolite formed from oxidative cleavage of the pyrrolidine ring at C5 followed by cyclisation of the aldehyde intermediate onto the benzimidazole moiety to form a stable 6-membered ring via M3, or via M5 shown in Figure 1.
Higher exposure to the 3 major metabolites was observed in Japanese versus Caucasian subjects, however due to the lower potency mentioned above, this observation is not thought to be of clinical importance. Furthermore, the metabolites are strong P-glycoprotein substrates and unlikely to cross the blood-brain barrier (Mühlan, 2021).
Covalent inactivation of CYP3A4
Futibatinib is primarily metabolised by CYP3A4 and to a lesser extent by CYP2C9 and CYP2D6; these alternative routes possibly being important given the inactivation of CYP3A4 by the acrylamide warhead of futibatinib itself.
Covalent modification of CYP3A4 is proposed to be due to metabolic activation of the acrylamide warhead to a highly electrophilic epoxide intermediate that covalently modifies CYP3A4 resulting in inactivation (Figure 2). The authors highlight that close scrutiny should be adopted when profiling the reactive metabolites of compounds possessing an α,β-unsaturated carbonyl scaffold, where epoxide formation could be a risk. (Tang et al., 2022).

Figure 2
Impact of inhibition of CYP3A4?
Vonoprazan is also highly metabolised. Unlike other Proton-pump inhibitors which are mainly metabolised through the polymorphic CYP2C19, vonoprazan is extensively biotransformed by CYP3A4 to inactive metabolites. There is only minor involvement of other CYPs such as CYP2D6 and CYP2C9, with additional conjugation by sulfotransferase SULT2. M-I and M-III are the major CYP3A4 metabolites (Figure 3) and glucuronides of these are subsequently formed (Sugano, 2018).
Since vonoprazan is mainly metabolised by CYP3A4, it is effective in inhibiting gastric acid in all patients without the variability in CYP2C19 metabolism across individuals. However, in the triple regimens to eradicate H. pylori infection, it is not clear how the coadministration of vonoprazan and clarithromycin, a CYP3A4 inhibitor, would impact routes of metabolism. It has also been shown that vonoprazan can inhibit CYP3A4, CYP2C9, CYP2D6, and CYP2B6, suggesting that the coadministration of vonoprazan and CYP substrates should be performed cautiously in clinical settings (Wang et al., 2020).

Figure 3: Main CYP3A4 metabolites of vonoprazan
CYP3A4 mediated pyrrole ring oxidation
Pactritinib is a multi-kinase JAK2 inhibitor used for the treatment of primary and secondary myelofibrosis approved by the FDA in 2022. It’s extensively biotransformed to 12 metabolites through multiple mechanisms; oxidation, N-dealkylation, O-dealkylation, hydrolysis, and dehydrogenation. CYP3A4 is involved in clearance to two major metabolites M1 and M2, representing 9.6% and 10.5% of parent drug exposure respectively. M1 results from oxidation of the pyrrole ring and M2 from O-dealkylation of the side chain.
Other identified metabolites include M3, resulting from a different oxidation in the pyrrole ring system, and M4 resulting from reduction of the double bond in the macrocyclic ring system. In human faeces M2 was the predominant metabolite (~24% of dose), whilst glucuronidation was observed to M7, the major metabolite in urine (~3% of dose).

Figure 3 Major CYP3A4 metabolites of pacritinib
Minor contribution from other CYPs
The 3β-methyl substitution on the neurosteroid ganaxalone is proposed to prevent rapid metabolism, although it is extensively metabolised by CYP3A4. A 16-OH metabolite generated via CYP3A4 metabolism has been identified as one of its major metabolites but other biotransformations have not yet been reported. There is minor involvement of other CYPs (CYP2B6, CYP2C19, CYP2D6).
Mitapivat is also extensively and primarily metabolised by CYP3A4, so much so, that hepatic impairment is expected to increase the systemic exposure of the drug. Minor contributions to metabolism from CYP1A2, CYP2C8 and CYP2C9 were observed during in vitro studies.
Similarly, olutasidenib is extensively biotransformed by CYP3A4 with the other 10% of dose metabolised through four other CYPs (CYP2C8, CYP2C9, CYP1A2, and CYP2C19). Pathways involved include N-dealkylation, demethylation, oxidative deamination followed by oxidation, and mono-oxidation with secondary glucuronidation.
Stay tuned for Part 2 of our blog on “Metabolism of 2022 FDA approved small molecule drugs”, where we look at metabolic routes of the other approved drugs where CYP3A4 is not the main driver of metabolism.
References
Iversen et al., 2022. Drug metabolism and drug transport of the 100 most prescribed oral drugs. Basic Clin Pharmacol Toxicol. 2022; 131(5): 311- 324. https://doi.org/10.1111/bcpt.13780
Maréchal et al., 2008. Insights into drug metabolism by cytochromes P450 from modelling studies of CYP2D6-drug interactions. Br J Pharmacol. 153 Suppl., S82-9. https://doi.org/10.1038/sj.bjp.0707570
Muehlan et al., 2019. Metabolism of the Dual Orexin Receptor Antagonist ACT-541468, Based on Microtracer/ Accelerator Mass Spectrometry. Curr Drug Metab. 20(4): 254-265. doi: 10.2174/1389200220666190206141814.
Mühlan, 2021. Dissertation at https://edoc.unibas.ch/83159/1/2021-05-28_Dissertation%20CM_final.pdf.
Sugano, 2018. Vonoprazan fumarate, a novel potassium competitive acid blocker, in the management of gastroesophageal reflux disease: Safety and clinical evidence to date. Therapeutic Advances in Gastroenterology. 2018;11. https://doi.org/10.1177/1756283X17745776
Tang et al., 2022. Covalent Inactivation of CYP3A by Futibatinib. Drug Metabolism and Disposition 50 (7) 931-941; DOI: https://doi.org/10.1124/dmd.122.000895
Wang et al., 2020. Cytochrome P450-Based Drug-Drug Interactions of Vonoprazan In Vitro and In Vivo. Front Pharmacol., 14; 11:53. https://doi.org/10.3389/fphar.2020.00053