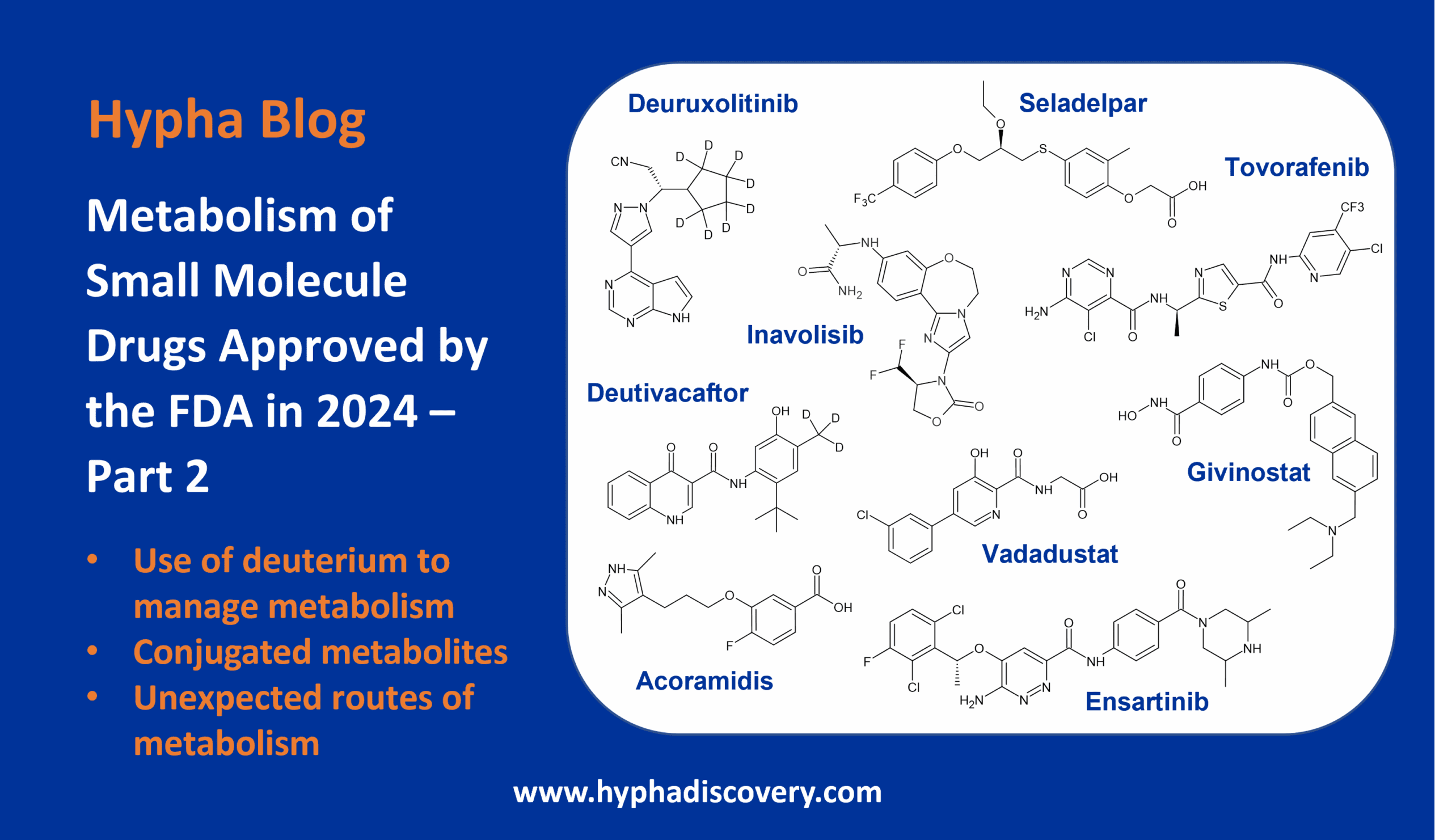
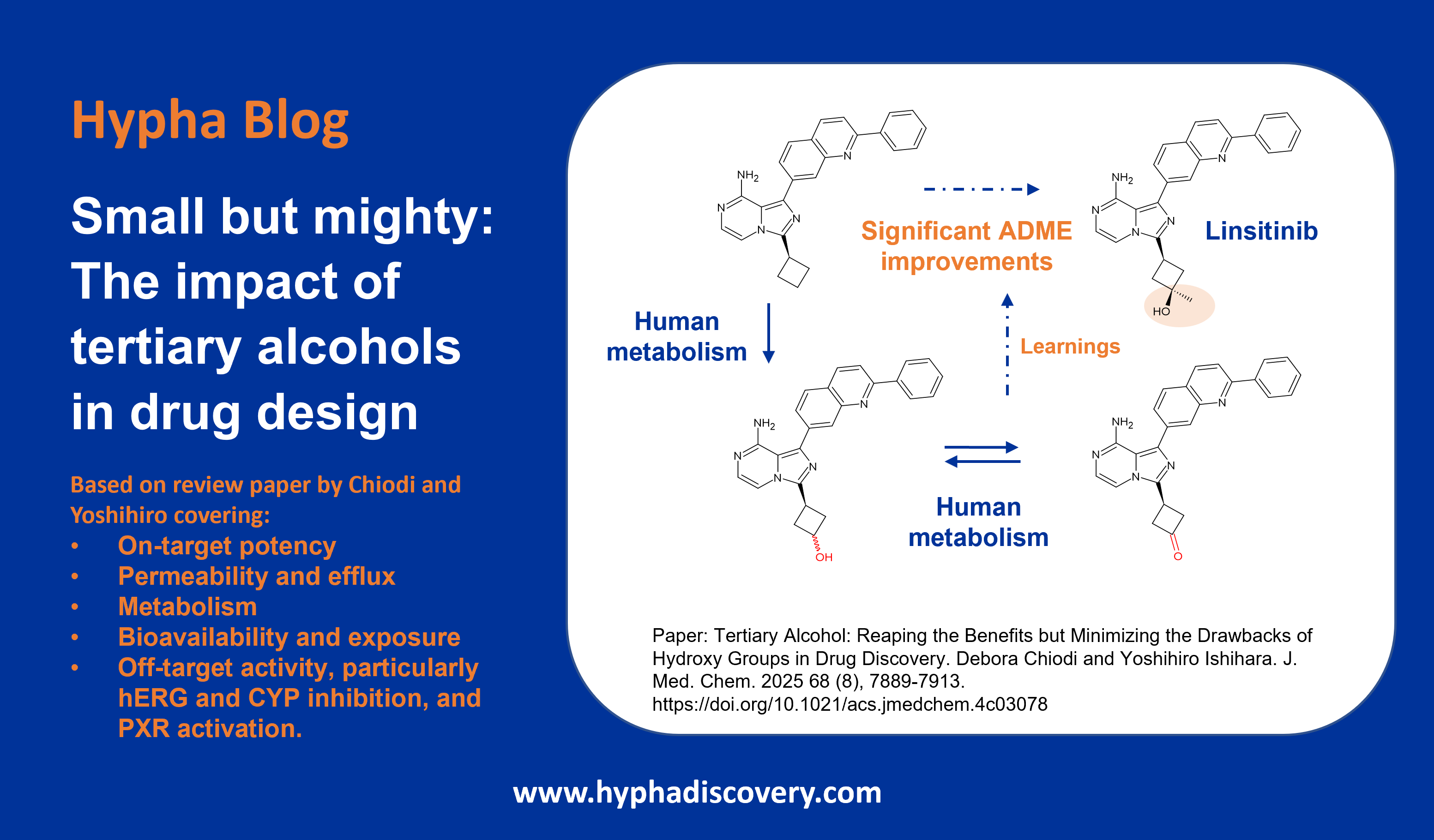
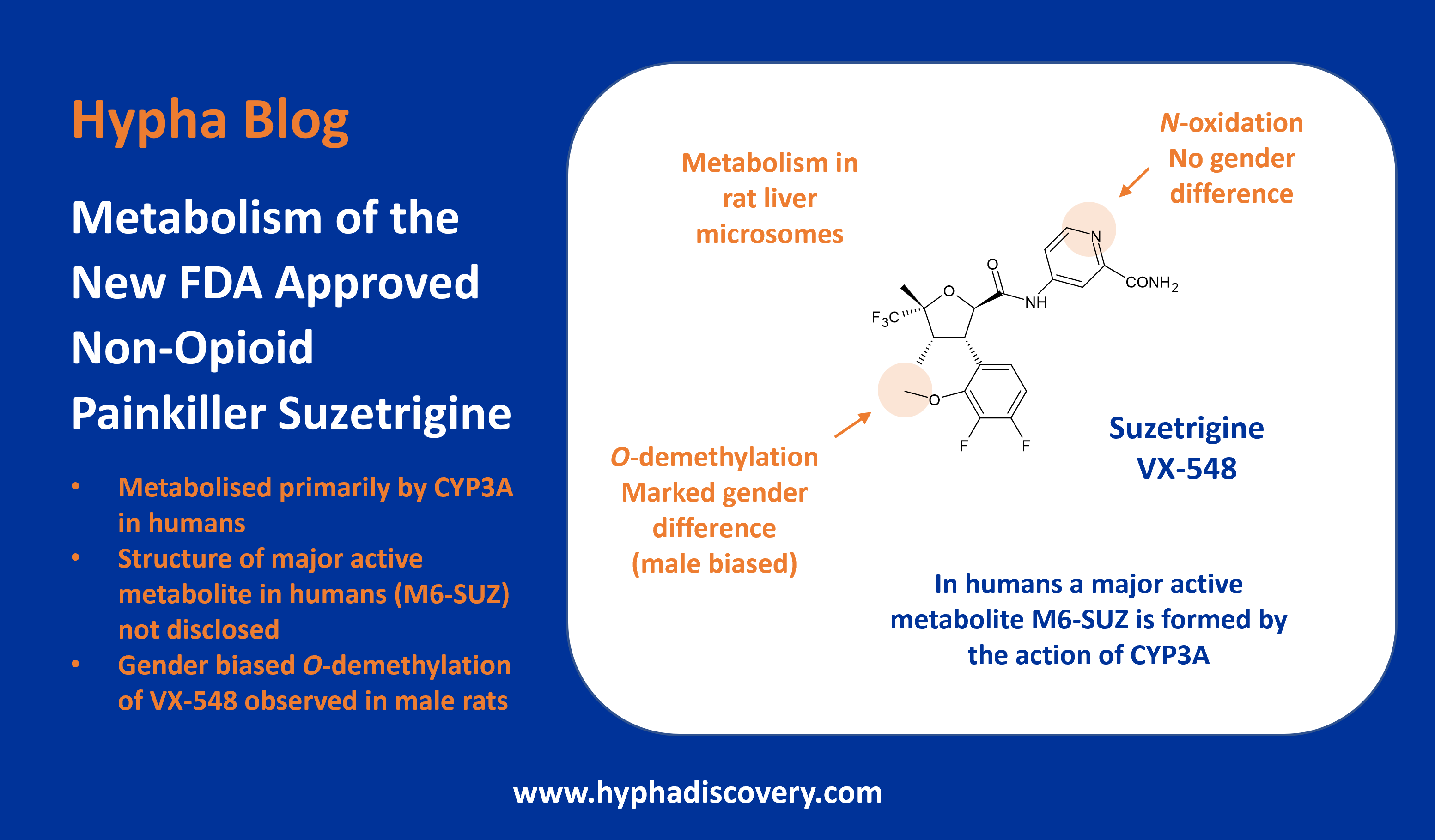
Metabolism of 2022 FDA approved small molecule drugs part 2
Mixing it Up
By Julia Shanu-Wilson
In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. In this follow-up blog, “Metabolism of 2022 FDA approved small molecule drugs part 2”, we dig into the other drugs in this set that are cleared by multiple CYPs, mixed phase mechanisms and through involvement of gut metabolism (see Table 1 at the end of this blog).
In humans, 90% of all drugs currently approved for clinical use are metabolised by one of seven CYP isoforms; CYP1A2, CYP2C8, CYP2C19, CYP2C19, CYP2D6, CYP2E1 and/or CYP3A4.1 Small molecule drugs approved in 2022 also adhere to this trend, with a couple also metabolised by CYP2B6 as part of a mixed CYP clearance mechanism.
The susceptibility of a drug to biotransformation by more than just one enzyme is a desirable property to minimise the risks from metabolism routed exclusively through polymorphic CYPs such as CYP2D6, CYP2C9 and CYP2C19, and to reduce the chance of drug-drug interactions. In SAR studies, structural modification of a lead compound mainly metabolised by a polymorphic enzyme could lead to the discovery of a new drug candidate which is metabolised by multiple enzymes.2
Involvement of multiple CYPs
Active Metabolites
The metabolism of Pfizer’s abrocitinib is mediated by multiple CYP enzymes to many metabolites, including two active metabolites with similar potency to the parent compound. Enzymes responsible involve CYP2C19 (~53%), CYP2C9 (~30%), CYP3A4 (~11%) and CYP2B6 (~6%).
Notable is the generation of 3 mono-hydroxylated metabolites, M1 (3-hydroxypropyl), M2 (2-hydroxypropyl) and M4 (pyrrolidinone pyrimidine), which circulate at >10% (Figure 1). At steady state, M2 and M4 are major metabolites whilst M1 is minor. Of the 3 metabolites in circulation, M1 and M2 have similar JAK inhibitory profiles as abrocitinib, while M4 is pharmacologically inactive. As a consequence, only ~60% of the pharmacologic activity of abrocitinib is attributable to the parent molecule with M1 and M2 contributing ~10% and ~30% respectively.3

Metabolism differences at steady state
Adagrasib, Mirati’s irreversible KRASG12C inhibitor for treatment of non-small cell lung cancer is mainly metabolised by CYP3A4. Interestingly however, since adagrasib (MRTX849) inhibits CYP3A4 following multiple dosing, its metabolism is subsequently taken over by other CYPs. In fact, CYP2C8, CYP1A2, CYP2B6, CYP2C9, and CYP2D6 all contribute to adagrasib’s metabolism at steady-state.
Oxidation of adagrasib occurs on the methylpyrrolidine group4. A poster presentation at the 2022 ISSX meeting on metabolism of MRTX849 revealed three circulating metabolites, M11, M55a and M68, at > 10% following a single oral dose. However only two of these, M11 and M68, were major metabolites at steady state through the involvement of CYP2C8 and other CYPs. Neither of these were human specific.5

Impact of CYP2C19 polymorphism
Despite mavacamten (Myokardia, now BMS) being highly metabolically stable, biotransformation results in multiple metabolites. In vitro studies indicated aromatic hydroxylation (M1), aliphatic hydroxylation (M2); N-dealkylation (M6), and glucuronidation of the M1-metabolite (M4) as major pathways.6 However, only CYP-mediated metabolism drives clearance in humans in vivo.
M1 is produced by para-hydroxylation of the phenyl-moiety primarily generated by CYP2C9, M2 is obtained from terminal hydroxylation of the isopropyl moiety and proposed to be mediated by CYP2C19, and M6 generated via CYP3A4/5-mediated N-dealkylation of the phenylethyl group. Concomitant use of mavacamten and drugs that interact with these enzymes may lead to life-threatening drug interactions such as heart failure or loss of effectiveness.7
Despite mavacamten’s extensive metabolism through multiple CYPs, it is largely driven by the polymorphic CYP2C19 (74%) with minor contributions from CYP3A4 (18%), and CYP2C9 (8%). In fact, mavacamten has a variable terminal half-life which depends on the CYP2C19 metabolic status of the patient, being 6-9 days in CYP2C19 normal metabolisers, but prolonged in CYP2C19 poor metabolisers to 23 days.7 Drug accumulation is thus dictated by the metabolism status for CYP2C19 with the largest accumulation observed in CYP2C19 poor metabolisers.

An aryl hydrocarbon receptor (AhR) agonist
Last year Consultant Andrew Parkinson wrote a piece for Hypha’s blog on AhR activators and whether they should be considered a friend or foe. Dermavant’s tapinarof is one such friend. Tapinarof is a topical aryl hydrocarbon receptor–modulating agent approved for the treatment of psoriasis by downregulating proinflammatory interleukin-17 and regulating expression of the skin-barrier proteins filaggrin and loricrin.8
This is not the only point of interest. Tapinarof (3,5-dihydroxy-4-isopropylstilbene) is actually a natural product which was found serendipitously when investigating the secondary metabolites of Photorhabdus luminescens, a bioluminescent, Gram-negative bacillus, which lives symbiotically within a soil-living entomopathogenic nematode. The worm carries the symbiont in its intestines and, upon entering a host insect, releases the bacilli, which help preserve insect tissue in optimal conditions for nematode growth. Remarkably, the mechanism of action of tapinarof was not determined until after profiling more than 800 potential cellular targets, where the most potent interactions were observed with AhR.
Tapinarof is metabolised by multiple pathways including oxidation, glucuronidation, and sulfation.9 In vivo, not unexpectedly, CYP1A2 and CYP3A4 are the major enzymes involved in hepatic metabolism, while CYP2C9, CYP2C19, and CYP2D6 play only minor roles.
Another active metabolite
Another approval for psoriasis is BMS’ oral drug deucravacitinib, which works via allosteric inhibition of TYK2 (tyrosine kinase 2). What makes deucravacitinib unique is its de novo design as a deuterated drug by the introduction of a deuterated methyl (CD3) amide group as a key pharmacophore. Deuteration at this position was found to block generation of a less selective primary amide metabolite in vivo and increase metabolic stability.
Despite deuteration, deucravacitinib still undergoes significant metabolism involving multiple pathways, involving CYP1A2, CYP2B6, CYP2D6, carboxylesterase (CES) 2, and uridine glucuronyl transferase (UGT) 1A9. N-demethylation of the triazole moiety in deucravacitinib by CYP1A2 results in a major metabolite BMT-153261, which has a comparable pharmacological activity to the parent drug and accounts for approximately 20% of the systemic exposure of the total drug-related components.10 Another major metabolite, BMT-158170, is generated by cyclopropyl carboxamide hydrolysis via CES2, however this is pharmacologically inactive. An N-glucuronide and a mono-oxidised metabolite are also formed.11
Involvement of phase II metabolism
Gilead’s lenacapavir is a long-acting capsid inhibitor for multi-drug resistant HIV and undergoes CYP3A4- and UGT1A1-mediated metabolism. Multiple mechanisms are at play covering oxidation, N-dealkylation, hydrogenation, amide hydrolysis, glucuronidation, hexose conjugation, pentose conjugation, and glutathione conjugation. However, CYP inhibitor studies point to glucuronidation being the primary route of metabolism. When coadministered with voriconazole, a CYP3A inhibitor, lenacapavir AUC was only increased 30%. However, when coadministered with atazanavir/cobicistat, a CYP3A/P-gp/UGT1A1 inhibitor, lenacapavir AUC increased 300%, suggesting that glucuronidation is the primary pathway for elimination.12
The metabolites of lenacapavir have not been fully characterized but no single circulating metabolite accounts for >10% of plasma drug-related exposure.13
Santen’s topical ocular drug omidenepag isopropyl is used to treat glaucoma where it is rapidly metabolised to the pharmacologically active form by CES 1 (carboxylesterase-1). Although CYP3A4 has been shown to play an important role in the subsequent metabolism of omidenepag in the liver, there are multiple routes at play. These include oxidation, N-dealkylation, glucuronidation, sulfate conjugation and, interestingly, taurine conjugation.14
Also involving conjugation is metabolism of taurursodiol (tauroursodeoxycholic acid), one of the two active components of Amylyx’s Relyvrio®, an oral treatment for amyotrophic lateral sclerosis (ALS). Taurursodiol is an ambiphilic bile acid and is the taurine conjugate of ursodiol. The major biotransformation seems to be deconjugation mediated by gut microbes, followed by reconjugation with glycine15, and thus ursodiol and glyco-ursodiol are observed as major metabolites of taurursodiol.16
This concludes our exploration of metabolism of last year’s FDA approved small molecule drugs. We hope it was a useful two-parter! We’re looking forward to the next crop!
Table 1 – Selected small molecule drugs approved by the FDA in 2022 with main reported routes of metabolism
 References for Metabolism of 2022 FDA approved small molecule drugs PART 2
References for Metabolism of 2022 FDA approved small molecule drugs PART 2
[1] Maréchal et al., 2008. Insights into drug metabolism by cytochromes P450 from modelling studies of CYP2D6-drug interactions. Br J Pharmacol. 2008 Mar;153 Suppl 1(Suppl 1):S82-9. https://doi.org/10.1038/sj.bjp.0707570
[2] Zhang and Tang, 2018. Drug metabolism in drug discovery and development. Acta Pharm Sin B. 2018 Sep;8(5):721-732. https://doi.org/10.1016/j.apsb.2018.04.003
[3] Bauman et al., 2022. The Pharmacokinetics, Metabolism, and Clearance Mechanisms of Abrocitinib, a Selective Janus Kinase Inhibitor, in Humans. Drug Metabolism and Disposition 50 (8) 1106-1118; DOI: https://doi.org/10.1124/dmd.122.000829
[4] Fell et al., 2020. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. Journal of Medicinal Chemistry 63 (13), 6679-6693. https://doi.org/10.1021/acs.jmedchem.9b02052
[5] Rahbaek et al., 2022. Adagrasib metabolism after single and multiple oral dosing in humans. Poster at ISSX/MDO 2022 Seattle meeting.
[6] Grillo et al., 2019. Adagrasib metabolism after single and multiple oral dosing in humans. Xenobiotica, 49:6, 718-733, https://doi.org/10.1080/00498254.2018.1495856
[7] FDA Prescribing information for CAMZYOS® (mavacamten) https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214998s000lbl.pdf
[8] Bissonnette et al., 2020. Tapinarof in the treatment of psoriasis: A review of the unique mechanism of action of a novel therapeutic aryl hydrocarbon receptor-modulating agent. J Am Acad Dermatol. 2021 Apr;84(4):1059-1067. https://doi.org/10.1016/j.jaad.2020.10.085
[9] FDA Prescribing information for VTAMA® (tapinarof) cream https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215272s000lbl.pdf
[10] FDA prescribing information for SOTYKTU® (deucravacitinib) https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/214958s000lbl.pdf
[11] BMS PRODUCT MONOGRAPH for SOTYKTU® (deucravacitinib) https://www.bms.com/assets/bms/ca/documents/productmonograph/SOTYKTU_EN_PM.pdf
[12] CLINICAL EVALUATION OF DRUG INTERACTIONS WITH ORAL LENACAPAVIR AND PROBE DRUGS. NATAP Conference presentation at https://www.natap.org/2021/CROI/croi_26.htm
[13] FDA prescribing information for SUNLENCA® (lenacapavir) https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215973s000lbl.pdf
[14] FDA prescribing information for OMLONTI® (omidenepag) https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215092s000lbl.pdf
[15] Setchell et al., 1996. Metabolism of orally administered tauroursodeoxycholic acid in patients with primary biliary cirrhosis. Gut 1996; 38: 439-446. http://dx.doi.org/10.1136/gut.38.3.439
[16] FDA prescribing information for RELYVRIO https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/216660s000lbledt.pdf