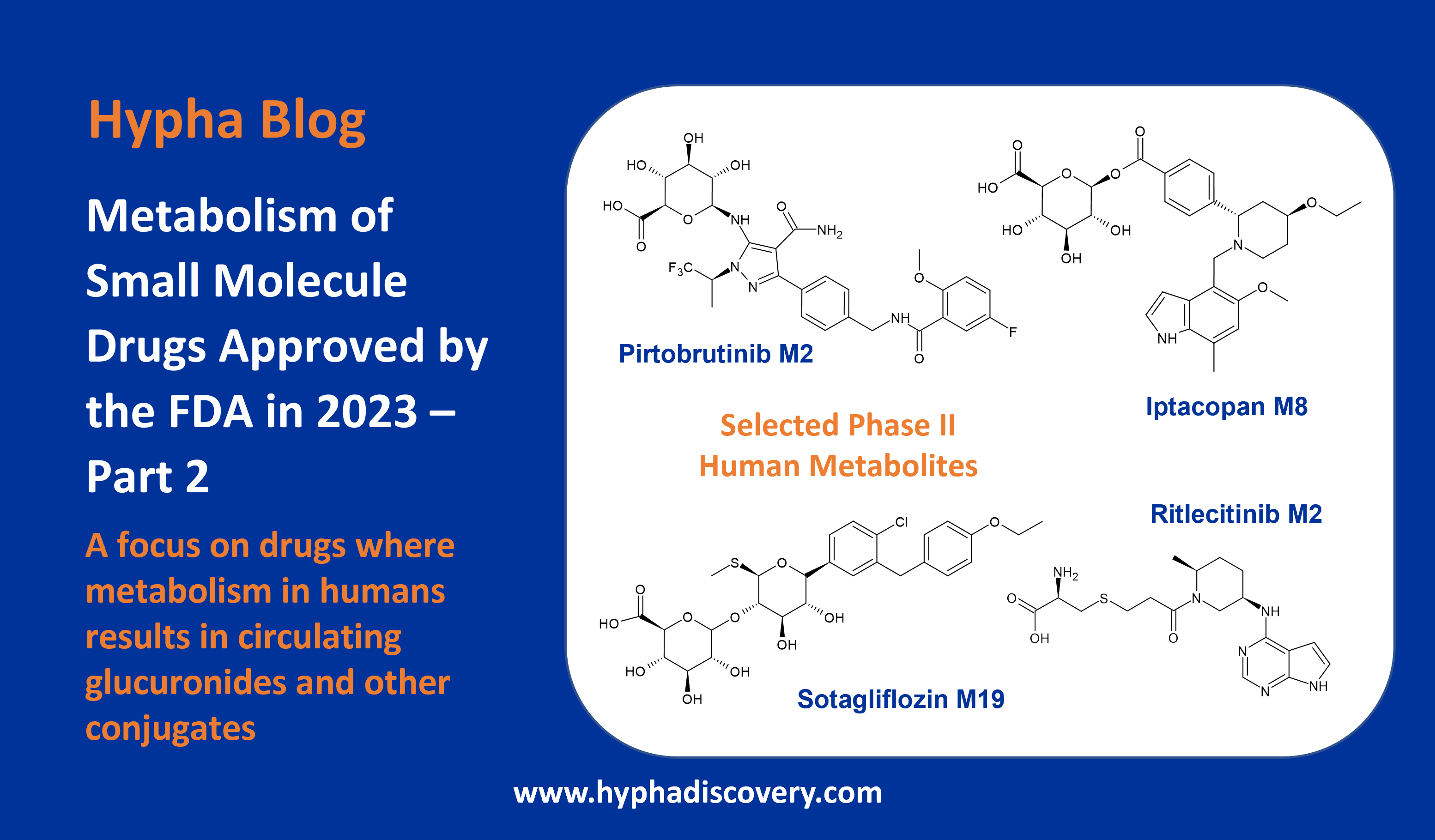
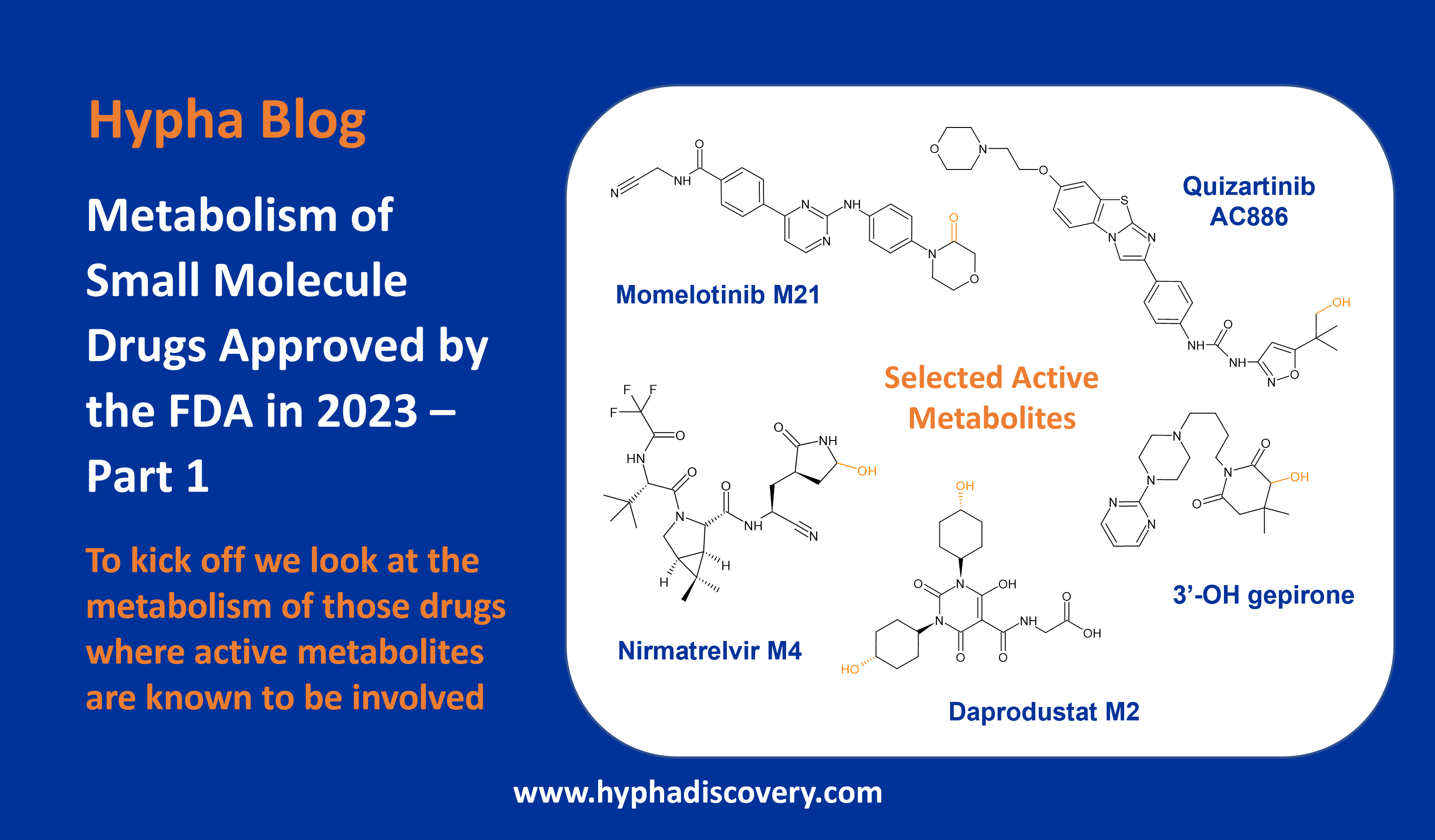
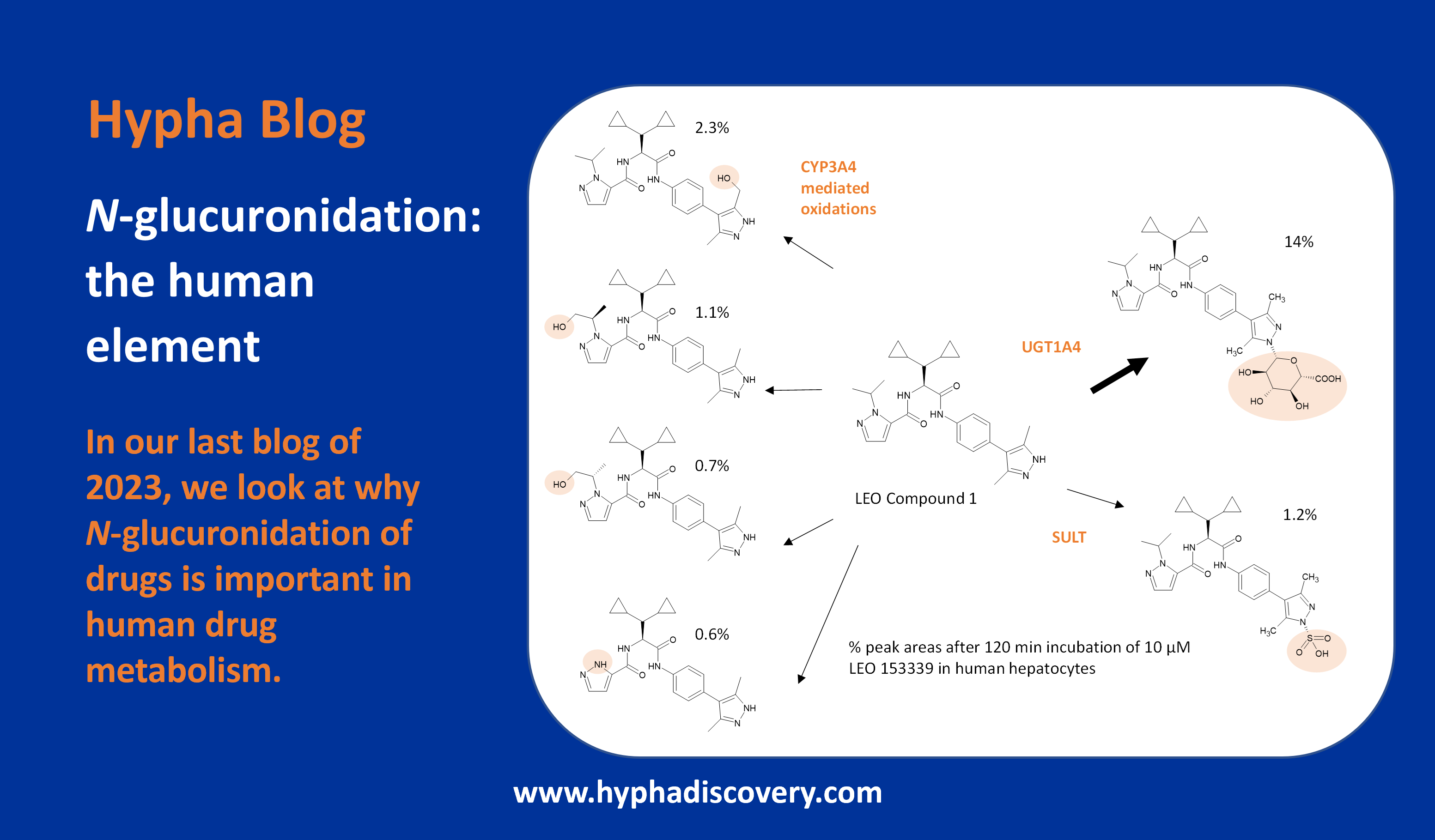
Orphan CYPs – finding a home in personalised medicine
By Samuel Coe
Cytochrome P450 enzymes (CYPs) are responsible for the metabolism of about 75% of marketed drugs. However, it is not the well-known workhorses of drug metabolism such as CYP3A4 that we want to focus on in this blog, but lesser known “orphan” CYPs.
Of the 57 CYPs identified by the human genome project, a great many have been denoted as “orphans” because their function, expression sites, and regulation are undefined. Since the expression of some of these orphan CYPs is tissue specific and increases in tumorigenesis, there is an excellent opportunity for the development of more targeted anti-cancer agents.
CYP activated prodrugs
Prodrugs are a class of molecule that are often inert on administration but undergo a metabolic change once administered releasing an active compound.
Several anti-cancer prodrugs, rely on metabolism by CYPs in the liver for their therapeutic action. For example, cyclophosphamide is activated in the liver by CYPs 2B6, 2C9 and 3A4 to form 4-hydroxycyclophosphamide.1 This hydroxylated metabolite is in equilibrium with its open ring tautomer, aldophosphamide (Figure 1). Both of these intermediate metabolites readily diffuse into cells but are not cytotoxic. Depending on the type of cell, aldophosphamide decomposes to form the cytotoxic phosphoramide mustard and the by-product acrolein via β-elimination. Once activated intracellularly, phosphoramide mustard can crosslink DNA, and in tumour cells this causes cell death. Unfortunately, cyclophosphamide is also highly cytotoxic to lymphocytes and a common side effect is leukopenia. Aldophosphamide is also oxidised to the inactive metabolite carboxyphosphamide by aldehyde dehydrogenase (ALDH).1

Figure 1 – Cyclophosphamide activation pathway adapted from Nat. Rev. Clin. Oncol. 6, 638–647 (2009)
There are steps being taken to reduce toxicity from circulating metabolites by co-administering CYP inhibitors, but in this blog article we highlight an alternative approach to CYP activated prodrugs that make use of a specific orphan CYP overly expressed in tumour cells to activate the prodrug, thus making it more target specific.
CYP2W1 – a tumour specific CYP
Completion of the human genome project in 2003 opened up possibilities for discovering new targets and insights into human diseases. One advancement since this milestone achievement has been the identification of previously unknown CYPs.2 This was done by cloning potential CYPs with close sequence homology to known families. Many new orphan CYPs have been cloned and characterised in CYP family 2, and CYP2W1 is of particular interest because of it’s potential as an oncology target. 2
The full CYP2W1 gene sequence was resolved in 2006 revealing closest sequence identities with CYP2D6 and CYP1A1, and is conserved across species.2 Although it is expressed in gastrointestinal tissues at the foetal stages, by 19 weeks in humans the gene is all but silenced, leaving its full function a mystery. Transcriptional regulation of the CYP2W1 gene is believed to be regulated by epigenetic methylation because the extent of methylation is inversely correlated with CYP2W1 expression levels.
The expression level of CYP2W1 is higher in colorectal cancer tumours. Two clinical studies using immunohistochemical staining of samples from colorectal cancer patients have shown that only 7% of tumours were not expressing CYP2W1, with the remaining 93% having weak, moderate or high staining. These results and others make a compelling case for CYP2W1 as a prognostic for colorectal cancer.
Targeting CYP2W1
Given CYP2W1 is essentially exclusively expressed in colorectal cancer tumour cells in adults, one potential approach is the development of a prodrug that becomes cytotoxic once metabolised specifically by CYP2W1. As you might imagine, with a target so potentially specific, efforts to identify scaffolds metabolised selectively by CYP2W1 have been undertaken in earnest.
Both truncated enzymes expressed in E. coli and full length CYP2W1 transfected into HEK293 cells have been shown to metabolise a wide variety of substrates (see Table 2 of reference 2), with indolines being the most relevant to this discussion.
The indoline-containing duocarmycins (Figure 2) are natural products produced by Streptomyces bacteria.3 Typified by two halves, a DNA binding subunit and a DNA alkylating subunit, they are antitumor antibiotics with the DNA alkylating activity derived from the ring-opening of a reactive cyclopropyl group.4
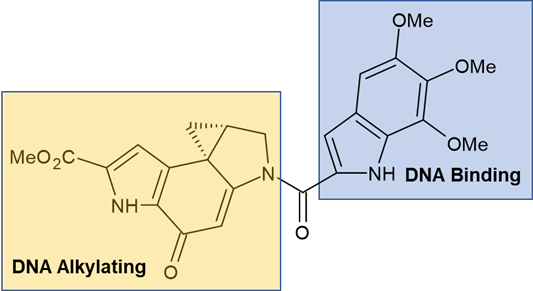
Figure 2 – Duocarmycin SA
Despite this family being some of the most potent tumour antibiotics ever discovered, this activity has led to problems in clinical development. Excessive toxicity in animal studies halted initial small molecule campaigns and subsequent attempts to balance stability and biological activity have not yet been successful.5
In spite of the challenges, one promising small molecule effort to capitalise on the potent activity of the duocarmycins are the chloromethylindoline analogues (Figure 3).6 These are promising prodrug scaffolds selectively activated by CYP2W1 and CYP1A1. Using the chloromethylindoline moiety as a masked cyclopropyl group, the key step is hydroxylation of the indoline. Hydroxylation by these CYPs activates a spiro-cyclisation cascade unveiling the cyclopropyl warhead. Subsequent ring opening by N3 of adenosine ultimately results in DNA replication stalling and cell death.6 A promising and elegant solution to a deadly cancer.
The clear advantage here is that the prodrug is completely benign prior to spirocyclisation, only becoming cytotoxic once already in the cancer cell, in stark contrast to cyclophosphamide mentioned earlier.
Unfortunately, due to the sequence homology with CYP1A1, selective activation by CYP2W1 is not yet been fully achieved, but there are hints that this could be developed in the future by varying the substitution of the DNA binding subunit.

Figure 3 – Chloromethylindoline pathway adapted from J. Med. Chem. 2013, 56, 6273−6277
Targeting CYP1A1
As we have seen when discussing CYP2W1, duocarmycins are also activated by CYP1A1. Other scaffolds have also been shown to be activated by CYP1A1 namely benzothiazoles, aminoflavones and phenyl 4-(2-oxo-3-alkylimidazolidin-1-yl)benzenesulfonates (PAIB-SOs). In fact, CYP1A1 has also been identified as one of the most promising targets for pro-drug activation due to its high expression in human breast cancer tumours.7
PAIB-SOs (Figure 4) have been shown to have low nanomolar cytotoxicity to a variety of breast cancer cell lines once N-dealkylated by CYP1A1.

Figure 4 – Showing the formation of activated PAIB-SOs
This dealkylation results in an active molecule that interferes with the interface of the α- and β-tubulins. Microtublins play pivotal roles in functions such as mitosis and cell mobility, but microtublin interfering agents have harmful effects such as bone marrow suppression as well as gastrointestinal and renal toxicity. Therefore, greater tumour specific activation is crucial to avoid potential side effects. Evidence of CYP selectivity has been shown against both isolated CYP3A4 and pooled human liver microsomes. A 4 h CYP3A4 incubation showed no turnover of the parent compound while a 4 h human liver microsome incubation showed only 3% turnover to the active compound with the major products being the parent and other unidentified oxidation products. It is still early stages for these compounds but these are excellent early indications that the PAIB-SOs might be successfully developed into breast cancer specific treatments.
Opportunities for other orphan CYPs
Besides CYP2W1, CYP4Z1 and CYP2S1 are frequently found to be upregulated in breast cancer. CYP4Z1 is an orphan CYP of particular interest as a target for preventing and treating breast cancer. It is known to form the signalling molecule 20-hydroxyeicosatetraenoic acid; however, it is the formation of 14,15-epoxyeicosatrienoic acid by CYP4Z1 that actually influences cellular proliferation, migration, and angiogenesis. Research to find a selective mechanism-based inhibitor is already underway.8
Due to its unique expression in all subtypes of breast cancer, CYP4Z1 could be exploited in prodrug design to increase specificity chemotherapy treatment. We hope to see the development of candidate prodrugs that can be activated by this enzyme in the not-too-distant future.
Personalised medicines of the future?
Drug discovery strategies based on the strategic use of the CYP family represent a promising approach for the development of new and highly selective tumour-targeting drugs. Above we have highlighted some of those exploiting CYP-dependent bioactivation of prodrugs. Such drugs will form part of what is dubbed “personalised medicines” and might in principle improve treatment regimens and efficacy while reducing the damaging effects associated with non-target drug action.
However, as we have seen this concept is difficult to implement, because success depends on the follow key parameters. Firstly, the CYP isoform selected for activating the prodrug must be overexpressed or inducible in the tumour, while remaining expressed at negligible levels in off-target tissues. Secondly, the design of the prodrug must aim for optimal ADMET properties to allow its efficient diffusion into the tumours, while its oxidation must release a cytocidal metabolite locally. Finally, such a prodrug must exhibit a high affinity and selectivity towards a specific CYP isoform, and the parent drug released upon enzymatic activation must bind its target(s) selectively and readily, with minimal entry into the general circulation.
With many small molecule prodrugs entering clinical trials it will be interesting to see which if any CYP activated assets can make it to market as specific and potent cancer treatments.
References
1Cyclophosphamide and cancer: golden anniversary. Emadi, A.; Jones, R. J.; Brodsky, R. A. Nat. Rev. Clin. Oncol., 2009, 6, 638–647
https://www.nature.com/articles/nrclinonc.2009.146 10.1038/nrclinonc.2009.146
2The CYP2W1 enzyme: regulation, properties and activation of prodrugs. Guo, J.; Johansson, I.; Mkrtchian, S.; Ingelman-Sundberg, M. Drug Metabolism Reviews., 2016, 48, 369-378
https://www.tandfonline.com/doi/full/10.1080/03602532.2016.1188939 10.1080/03602532.2016.1188939
3Duocarmycin A, a new antitumor antibiotic from Streptomyces. Takahashi, K.; Michio, I.; Makoto, M.; Kozo, A.; Isao, K.; Fusao, T.; Hirofumi, N. The Journal of Antibiotics., 1988, 41, 1915-1917
https://www.jstage.jst.go.jp/article/antibiotics1968/41/12/41_12_1915/_article 10.7164/antibiotics.41.1915
4CC-1065 and the duocarmycins: unraveling the keys to a new class of naturally derived DNA alkylating agents. Boger, D. L.; Johnson, D. S. Proc. Natl. Acad. Sci. USA., 1995, 92, 3642-3649
https://www.pnas.org/doi/abs/10.1073/pnas.92.9.3642 10.1073/pnas.92.9.3642
5Chemical and Biological Explorations of the Family of CC-1065 and the Duocarmycin Natural Products. Ghosh, N.; Sheldrake, H. M.; Searcey, M.; Pors, K. Current Topics in Medicinal Chemistry, 2009, 9, 1494-1524
http://www.eurekaselect.com/article/30324 10.2174/156802609789909812
6Re-engineering of the Duocarmycin Structural Architecture Enables Bioprecursor Development Targeting CYP1A1 and CYP2W1 for Biological Activity. Sheldrake, H. M.; Travica, S.; Johansson, I.; Loadman, P. M.; Sutherland, M.; Elsalem, L.; Illingworth, N.; Cresswell, A. J.; Reuillon, T.; Shnyder, S. D.; Mkrtchian, S.; Searcey, M.; Ingelman-Sundberg, M,.; Patterson, L. H.; Pors, K. J. Med. Chem. 2013, 56, 6273−6277
https://pubs.acs.org/doi/10.1021/jm4000209 10.1021/jm4000209 10.1021/jm4000209
7Activation of Phenyl 4-(2-Oxo-3-alkylimidazolidin-1-yl)benzenesulfonates Prodrugs by CYP1A1 as New Antimitotics Targeting Breast Cancer Cells. Fortin, S.; Charest-Morin, X.; Turcotte, V.; Lauvaux, C.; Lacroix, J.; Côté, M-F.; Gobeil, S.; Gaudreault, R. C. J. Med. Chem. 2017, 60, 4963-4982
https://pubs.acs.org/doi/10.1021/acs.jmedchem.7b00343 10.1021/acs.jmedchem.7b00343
8What might the presence of ‘orphan’ CYP450 isoforms in breast epithelial cells mean for the future of targeted therapeutics? Licznerska, B; Baer-Dubowska, W. Expert Opinion on Drug Metabolism & Toxicology, 2021, 17:2, 135-137
https://www.tandfonline.com/doi/full/10.1080/17425255.2021.1844182