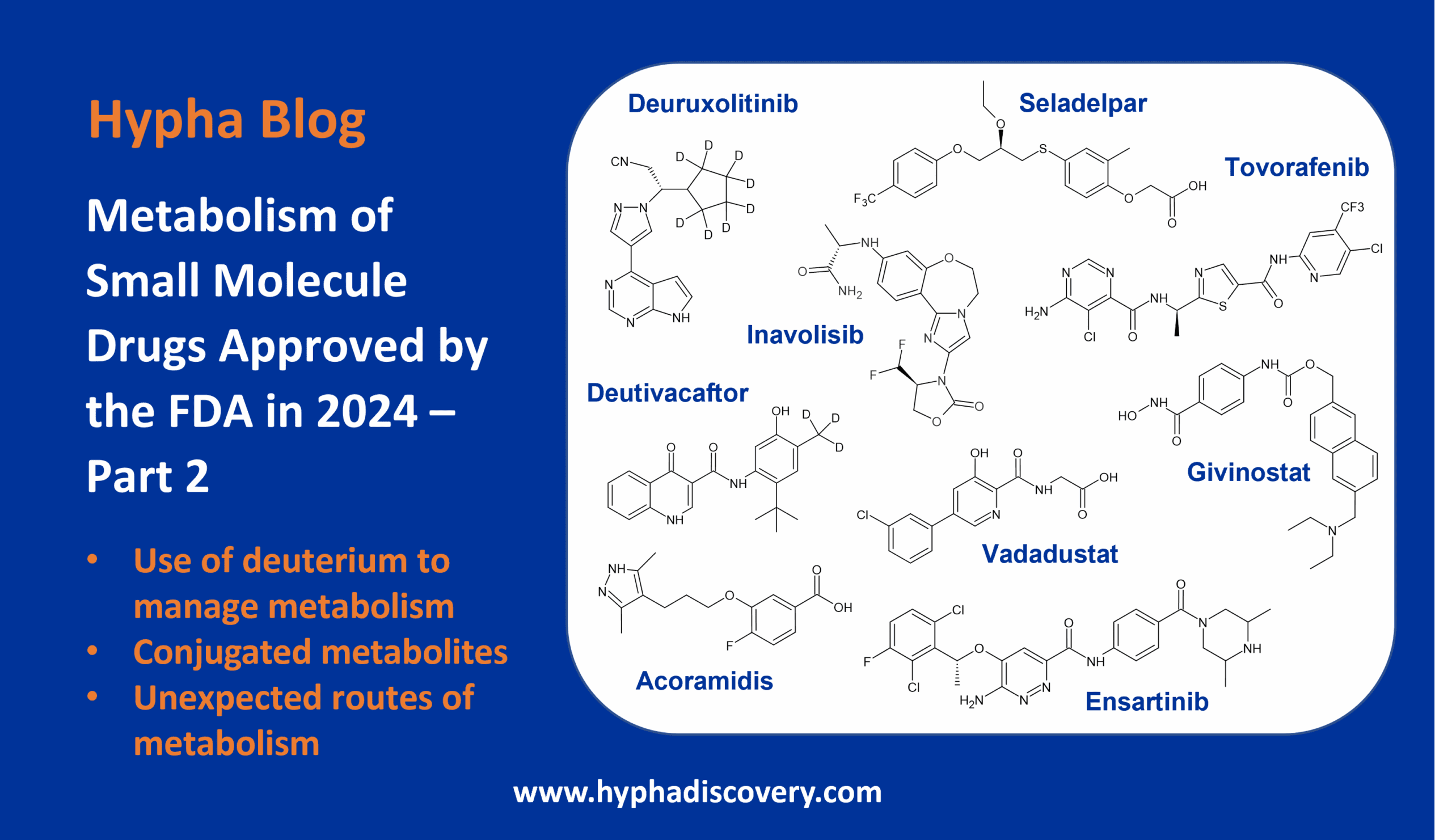
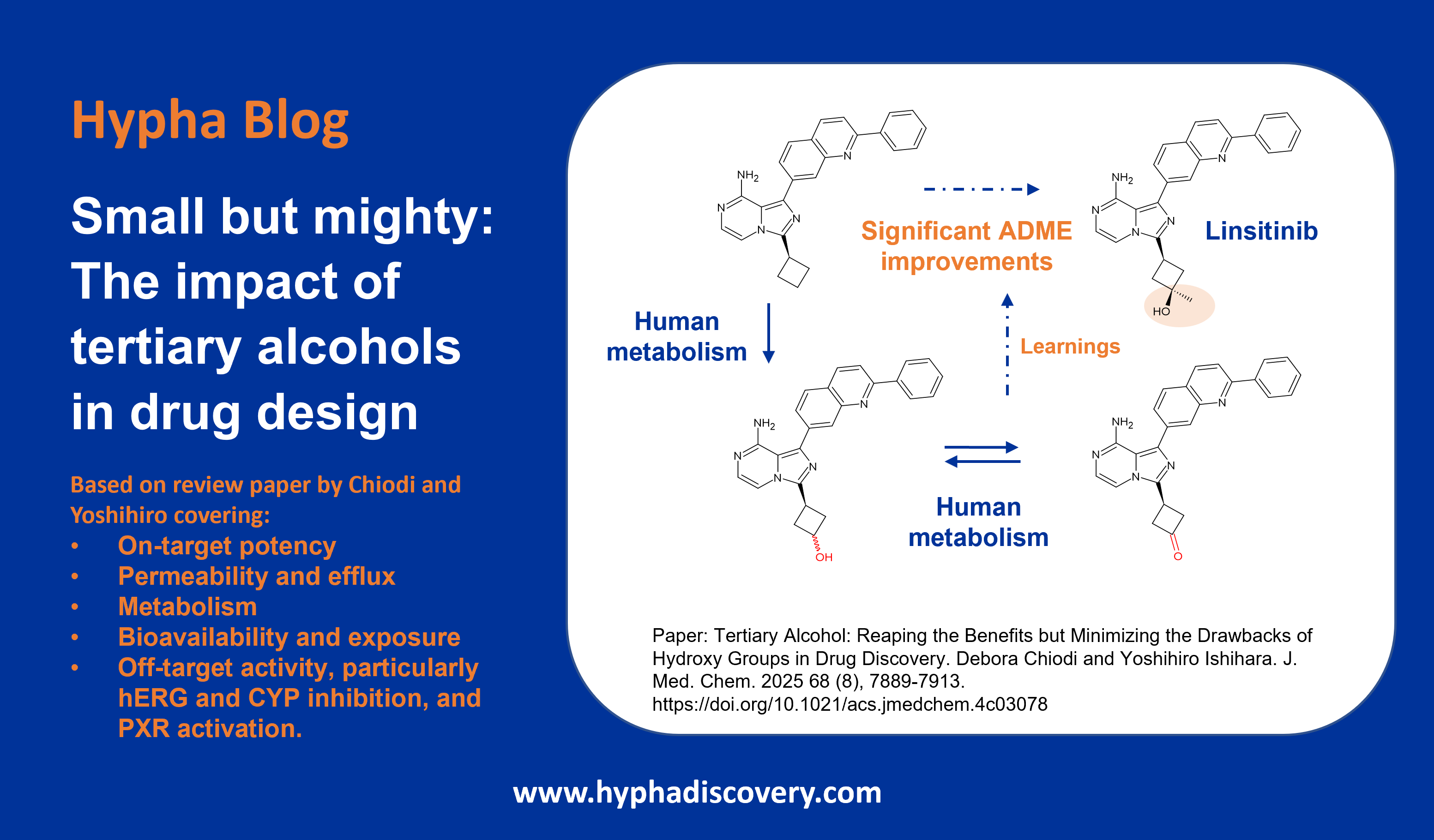
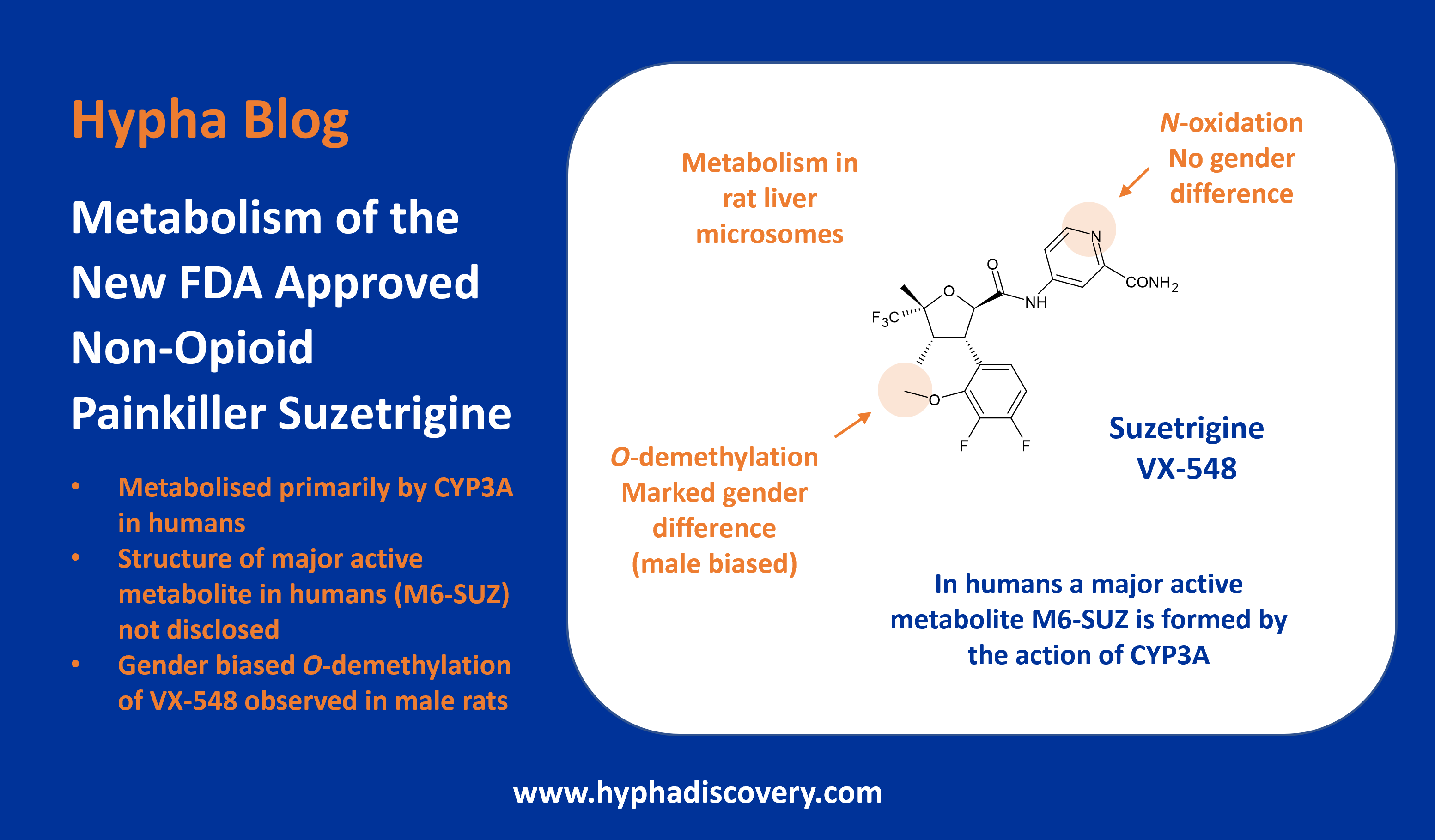
Metabolism of t-butyl groups in drugs: t for trouble?
By Julia Shanu-Wilson
The tert-butyl group is a common group present in many drugs across different therapeutic classes. Despite its simple bulky structure, t-butyl groups have unique properties1 and can serve to change or increase specificity, or act as steric shields to increase stability of compounds containing chemically or enzymatically susceptible groups.2 However, they are often subject to metabolism by a number of cytochrome P450 enzymes (CYPs).
Involvement of different CYPs
In ombitasvir, an antiviral drug for the treatment of hepatitis C, inclusion of a t-butyl group provides a potency enhancement,3 and in nelfinavir, an antiretroviral drug used in the treatment of HIV, the t-butylcarboxamide moiety nicely occupies a subsite of HIV-1 protease.4 However, these t-butyl groups are targeted by CYPs.
Following enzymatic amide hydrolysis of ombitasvir to M23, oxidation at the t-butyl position by CYP2C8 results in a family of metabolites via M26. Oxidation and C-demethylation of M26 results in 7 metabolites, as well as 2 other metabolites arising from oxidation of M26. M29 and M36 are the major human circulating metabolites.5

In contrast, the t-butyl group of nelfinavir is hydroxylated by the polymorphic CYP2C19 to the main human metabolite M8.6 M8 displays potent anti-viral activity. Interestingly M8 binds to human pregnane X receptor (PXR) and act as a partial agonist and competitive antagonist in the same way as nelfinavir, which is being investigated for repurposing as an anticancer medication.7

Another anti-viral drug where hydroxylation at the t-butyl group results in an active metabolite is dasabuvir. Oxidation to M1 is followed by glucuronidation, sulfation and additional secondary oxidation to the t-butyl-acid M5. Like ombitasvir, CYP2C8 is also the main enzyme involved in its metabolism.8

Drug Hunter’s Molecule of the Year for 2021, Pfizer’s COVID-19 drug nirmatrelvir (Paxlovid) contains a t-butyl group. Here, metabolism of the drug is largely driven by CYP3A4 and in fact, the drug is co-administered with the CYP3A4 inhibitor ritonavir to slow metabolism. However, in Paxlovid, the major metabolite is derived from pyrrolidinone ring oxidation with t-butyl oxidation to M3 only a more minor contribution. M4 demonstrated potent inhibition of recombinant SARS-CoV-2 3CLpro activity, but was relatively weaker as an antiviral agent in cells, potentially due to reduced permeability.9

A metabolite as a better drug
The tale of terfenadine and fexofenadine is a classic story. Terfenadine is an antihistamine drug (long withdrawn from the market) which caused QT interval prolongation. However, oxidation of terfenadine’s t-butyl group to an acid by CYP3A4 results in the active metabolite fexofenadine, which has negligible cardiotoxicity.

Strategies to reduce clearance
Since susceptibility to metabolism by CYPs is a common issue with t-butyl group in compounds of interest to medicinal chemists, what else can be done?
Substitution
Barnes-Seeman et al., suggest polar substitution such as a hydroxyl, cyano or acid group is a possibility if tolerated by the receptor site, since this substitution can increase metabolic stability. If such a substitution is not tolerated, as an alternative they designed a replacement group combining a cyclopropyl and a trifluoromethyl (Cp- CF3) group. This lacks fully sp3 C-Hs, thus lowering the rate of H-abstraction by CYP enzymes. Installation of such a Cp- CF3 group significantly increased metabolic stability in their compound.10
An (obvious) word of caution though, replacing t-butyl with Cp-CF3 does not reduce metabolism at distal soft spots, and so metabolically liability can be deflected to another area in the molecule. The t-butyl group in the antiandrogen finasteride is first hydroxylated by CYP3A4 to M1 then by aldehyde dehydrogenase via an aldehyde intermediate to an acid group in M3. These are the two major metabolites in humans, possessing 20% of the inhibitory activity of finasteride.11 Swapping the t-butyl group with Cp- CF3 causes only a moderate increase in metabolic stability in human liver microsomes, since metabolism at the 6 position to M4 is still possible.10

The t-butyl group of the endothelin receptor antagonist bosentan is hydroxylated to a major active metabolite by polymorphic CYP2C9.12 CYP3A4 also plays a role in its metabolism through demethylation. In fact, the t-butyl hydroxylation is used as a control reaction in Hypha’s PolyCYPs metabolite screening kits, since bosentan is included as a control substrate in the kits. Onward oxidation to the carboxyl is also observed at the t-butyl position.

Efforts to divert metabolism away from CYP2C9 by replacing bosentan’s t-butyl group with perfluorinated bioisoteres impeded the specific CYP2C9 driven hydroxylation reaction, however this did not lead to increased metabolic stability due to metabolic compensation elsewhere, notably O-demethylation reactions. A MetaSite design led approach also resulted in a compound stable to CYP2C9 but unstable in human liver microsomes.13
Deuteration
It would be remiss to not mention Vertex’s Ivacaftor in a blog about t-butyl groups since this drug contains two of these motifs. The t-butyl at the 4-position boosts potency whereas the other t-butyl at the 2-position adds lipophilic character at the para position of the phenol ring along with an improved pk profile.14
One of the t-butyl groups of Ivacaftor is metabolized by CYP3A4 to a hydroxylated active metabolite M1, with a potency 1/6th that of Ivacaftor itself, which is then further oxidized to the acid group in M6, the latter having no appreciable activity. Both are major metabolites in humans.15
To reduce clearance, Concert Pharmaceuticals developed a deuterated form of Ivacaftor, CTP-656, which has since been acquired by Vertex and is in phase 3 clinical trials as VX-561 (Deutivacaftor) in combination therapy for treatment of cystic fibrosis. The deuterated form of the drug has greater metabolic stability and thus a reduced rate of clearance and longer half-life. Fortunately there was no metabolic switching effect with CTP-656, only production of reduced levels of the same plasma metabolites as Ivacaftor. 16

 Sometimes the effect of deuterium substitution can be unpredictable, as seen with another deuterated analogue – d18-Ivacaftor – that was being evaluated at the same time as CTP-656. In fact, in an animal study d18-Ivacaftor showed increased exposure versus Ivacaftor in one species but not the other.16
Sometimes the effect of deuterium substitution can be unpredictable, as seen with another deuterated analogue – d18-Ivacaftor – that was being evaluated at the same time as CTP-656. In fact, in an animal study d18-Ivacaftor showed increased exposure versus Ivacaftor in one species but not the other.16
So even though slower metabolism is clear in vitro, the translation to in vivo does not necessarily follow since other clearance mechanisms may predominate, and may vary between species.

The team at Astex Pharmaceuticals UK and Cancer Research UK Newcastle Drug Discovery Unit also applied deuterium to some novel isoindolinone inhibitors of the p53-MDM2 protein-protein interaction.17 This alleviated oxidation at a hydroxymethylcyclopropyl chain adjacent to the terminal hydroxyl group, as well as reducing intestinal CYP3A4 metabolism to maximize oral bioavailability.
Also interesting was the early replacement of a methyl of the other t-butyl group with a hydroxyl group which maintained the desired Try67 OUT conformation while forming a favorable H-bond with an amino acid residue Gln72. As a bonus, the substitution also introduced a less lipophilic and more drug-like substituent into the molecule.
Not always trouble
Whilst susceptibility of t-butyl groups to CYP mediated metabolism can pose a problem, this is not always the case. If there are issues with high clearance as a result, different strategies can be tried to circumvent this, bearing in mind that a switch out from one route of metabolism can often redirect to other metabolically labile sites in the compound.
References
1 The tert-butyl group in chemistry and biology. Philippe Bisel, Loay Al-Momanib and Michael Müller. Org. Biomol. Chem., 2008,6, 2655-2665; DOI: https://doi.org/10.1039/B800083B
2 An Introduction to Medicinal Chemistry. Graham L. Patrick. Oxford University Press, 2017, p259. ISBN: 9780198749691
3 Discovery of ABT-267, a Pan-Genotypic Inhibitor of HCV NS5A. David A. DeGoey, John T. Randolph, Dachun Liu, John Pratt, Charles Hutchins, Pamela Donner, A. Chris Krueger, Mark Matulenko, Sachin Patel, Christopher E. Motter, Lissa Nelson, Ryan Keddy, Michael Tufano, Daniel D. Caspi, Preethi Krishnan, Neeta Mistry, Gennadiy Koev, Thomas J. Reisch, Rubina Mondal, Tami Pilot-Matias, Yi Gao, David W. A. Beno, Clarence J. Maring, Akhter Molla, Emily Dumas, Andrew Campbell, Laura Williams, Christine Collins, Rolf Wagner, and Warren M. Kati. J. Med. Chem. 2014, 57, 5, 2047–2057; https://doi.org/10.1021/jm401398x
4 Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease. Stephen W. Kaldor, Vincent J. Kalish, Jay F. Davies, Bhasker V. Shetty, James E. Fritz, Krzysztof Appelt, Jeffrey A. Burgess, Kristina M. Campanale, Nickolay Y. Chirgadze, David K. Clawson, Bruce A. Dressman, Steven D. Hatch, Deborah A. Khalil, Maha B. Kosa, Penny P. Lubbehusen, Mark A. Muesing, Amy K. Patick, Siegfried H. Reich, Kenneth S. Su, and John H. Tatlock. J. Med. Chem. 1997, 40, 24, 3979–3985; https://doi.org/10.1021/jm9704098
5 Metabolism and Disposition of [14C]Ombitasvir in Humans. Jianwei Shen, Michael Serby, Bruce Surber, Anthony J. Lee, Junli Ma, Prajakta Badri, Rajeev Menon, Olga Kavetskaia, Sonia M. de Morais, Jens Sydor and Volker Fischer. Drug Metabolism and Disposition August 1, 2016, 44 (8) 1148-1157; DOI: https://doi.org/10.1124/dmd.115.067496
6 Conversion of the HIV protease inhibitor nelfinavir to a bioactive metabolite by human liver CYP2C19. Vandana N. Hirani, Judy L. Raucy and Jerome M. Lasker. Drug Metabolism and Disposition December 2004, 32 (12) 1462-1467; DOI: https://doi.org/10.1124/dmd.104.001743
7 Nelfinavir and its active metabolite M8 are partial agonists and competitive antagonists of the human pregnane X receptor. Oliver Burk, Thales Kronenberger, Oliver Keminer, Serene M. L. Lee, Tobias S. Schiergens, Matthias Schwab and Björn Windshügel. Molecular Pharmacology March 2021, 99 (3) 184-196; DOI: https://doi.org/10.1124/molpharm.120.000116
8 Metabolism and Disposition of Hepatitis C Polymerase Inhibitor Dasabuvir in Humans. Jianwei Shen, Michael Serby, Aimee Reed, Anthony J. Lee, Rajeev Menon, Xiaomei Zhang, Kennan Marsh, Xia Wan, Olga Kavetskaia and Volker Fischer. Drug Metabolism and Disposition August 2016, 44 (8) 1139-1147; DOI: https://doi.org/10.1124/dmd.115.067512
9 Disposition of Nirmatrelvir, an Orally Bioavailable Inhibitor of SARS-CoV-2 3C-Like Protease, across Animals and Humans. Heather Eng, Alyssa L. Dantonio, Eugene P. Kadar, R. Scott Obach, Li Di, Jian Lin, Nandini C. Patel, Britton Boras, Gregory S. Walker, Jonathan J. Novak, Emi Kimoto, Ravi Shankar P. Singh and Amit S. Kalgutkar. Drug Metabolism and Disposition May 2022, 50 (5) 576-590; DOI: https://doi.org/10.1124/dmd.121.000801
10 Metabolically Stable tert-Butyl Replacement. David Barnes-Seeman, Monish Jain, Leslie Bell, Suzie Ferreira, Scott Cohen, Xiao-Hui Chen, Jakal Amin, Brad Snodgrass, and Panos Hatsis. ACS Med. Chem. Lett. 2013, 4, 6, 514–516; https://doi.org/10.1021/ml400045j
11 Identification of Finasteride Metabolites in Human Bile and Urine by High-Performance Liquid Chromatography/Tandem Mass Spectrometry. Anna Lundahl, Hans Lennernäs, Lars Knutson, Ulf Bondesson and Mikael Hedeland. Drug Metabolism and Disposition October 2009, 37 (10) 2008-2017; DOI: https://doi.org/10.1124/dmd.109.027870
12 Absorption, Excretion, and Metabolism of the Endothelin Receptor Antagonist Bosentan in Healthy Male Subjects. Cornelia Weber, Rodolfo Gasser and G. Hopfgartner. Drug Metabolism and Disposition July 1999, 27 (7) 810-815.
13 Metabolism study and biological evaluation of bosentan derivatives. Susan Lepri, Laura Goracci, Aurora Valeri, Gabriele Cruciani. European Journal of Medicinal Chemistry, Volume 121, 4 October 2016, Pages 658-670; https://doi.org/10.1016/j.ejmech.2016.06.006
14 Discovery of N-(2,4-Di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, Ivacaftor), a Potent and Orally Bioavailable CFTR Potentiator. Sabine Hadida, Fredrick Van Goor, Jinglan Zhou, Vijayalaksmi Arumugam, Jason McCartney, Anna Hazlewood, Caroline Decker, Paul Negulescu, and Peter D. J. Grootenhuis. J. Med. Chem. 2014, 57, 23, 9776–9795; https://doi.org/10.1021/jm5012808
15 PharmGKB summary ivacaftor pathway, pharmacokinetics/pharmacodynamics. Fohner, Alison E.; McDonagh, Ellen M.; Clancy, John P.; Whirl Carrillo, Michelle; Altman, Russ B.; Klein, Teri E. Pharmacogenetics and Genomics: January 2017, Volume 27 (1), p 39-42; doi: 10.1097/FPC.0000000000000246
16 Altering Metabolic Profiles of Drugs by Precision Deuteration 2: Discovery of a Deuterated Analog of Ivacaftor With Differentiated Pharmacokinetics for Clinical Development. Scott L. Harbeson, Adam J. Morgan, Julie F. Liu, Ara M. Aslanian, Sophia Nguyen, Gary W. Bridson, Christopher L. Brummel, Lijun Wu, Roger D. Tung, Lana Pilja, Virginia Braman and Vinita Uttamsingh. Journal of Pharmacology and Experimental Therapeutics June 13, 2017, jpet.117.241497; DOI: https://doi.org/10.1124/jpet.117.241497
17 Structure-Based Design of Potent and Orally Active Isoindolinone Inhibitors of MDM2-p53 Protein–Protein Interaction. Gianni Chessari, Ian R. Hardcastle, Jong Sook Ahn, Burcu Anil, Elizabeth Anscombe, Ruth H. Bawn, Luke D. Bevan, Timothy J. Blackburn, Ildiko Buck, Celine Cano, Benoit Carbain, Juan Castro, Ben Cons, Sarah J. Cully, Jane A. Endicott, Lynsey Fazal, Bernard T. Golding, Roger J. Griffin, Karen Haggerty, Suzannah J. Harnor, Keisha Hearn, Stephen Hobson, Rhian S. Holvey, Steven Howard, Claire E. Jennings, Christopher N. Johnson, John Lunec, Duncan C. Miller, David R. Newell, Martin E. M. Noble, Judith Reeks, Charlotte H. Revill, Christiane Riedinger, Jeffrey D. St. Denis, Emiliano Tamanini, Huw Thomas, Neil T. Thompson, Mladen Vinković, Stephen R. Wedge, Pamela A. Williams, Nicola E. Wilsher, Bian Zhang, and Yan Zhao. J. Med. Chem. 2021, 64, 7, 4071–4088; https://doi.org/10.1021/acs.jmedchem.0c02188