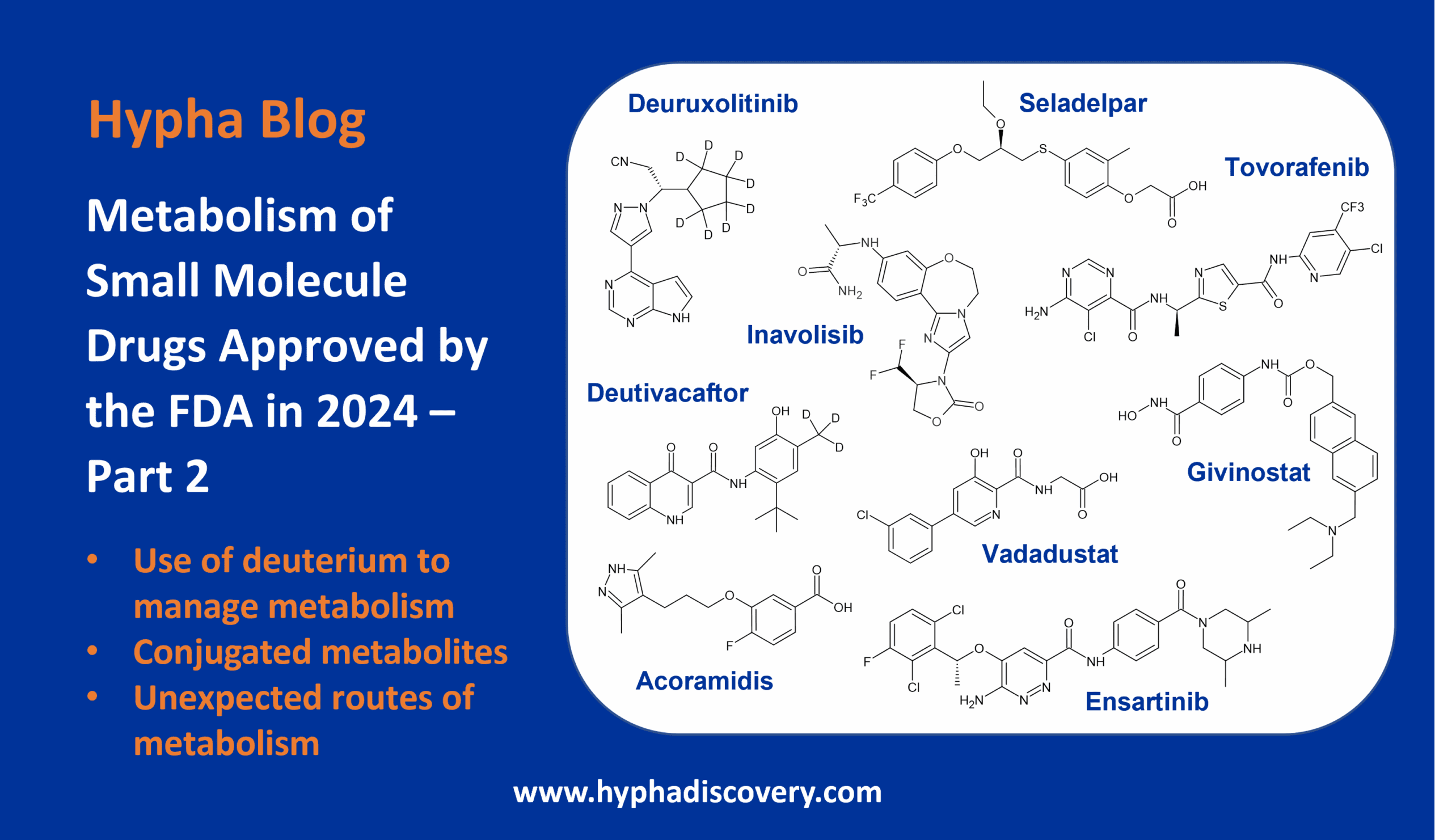
N-glucuronidation: the human element
By Julia Shanu-Wilson
In our last blog of the year, we look at why N-glucuronidation of drugs is important in human drug metabolism.
Glucuronidation is the most common phase II reaction observed in the metabolism of drugs in humans. Conjugation of small molecule drugs to glucuronic acid is catalysed by several UGTs to frequently form N– and O-glucuronides. Relative to the parent drug, glucuronides are more polar and rely more on active transport for movement across membranes, thus raising the probability of transporter-mediated interactions. The development of less lipophilic drugs with reduced CYP metabolism heightens the potential for metabolism through phase II metabolic routes and consequently involvement of transporters. Thus, although phase II metabolites are invariably pharmacologically inactive (with a few notable exceptions), a proactive approach to exploring the transport properties of major glucuronides is recommended in order to interrogate any DDI liabilities [1].
N-glucuronidation and involvement of UGT1A4
N-glucuronides can be formed as a result of aliphatic and aromatic conjugations with pyrazole, pyridine, pyridazine, pyrrolidine, pyrimidine, imidazole, triazole, and tetrazole moieties all being susceptible ring structures. At Hypha we have seen N-glucuronides formed at most of these moieties in drug structures, and of note, N-glucuronidation of pyrazoles.
The pyrazole in a drug compound developed by LEO as an oral IL-17A protein-protein interaction modulator for the treatment of psoriasis and other inflammatory disorders is susceptible to N-glucuronidation. The drug is metabolised through multiple phase I and phase II routes, but with N-glucuronidation in the pyrazole moiety resulting in a major metabolite in humans via the action of UGT1A4. Only very small amounts of the N-glucuronide were observed in other species. In fact the interspecies variability in N-glucuronidation is particularly high, especially for aliphatic tertiary amines and, relevant to the LEO drug compound, aromatic N-heterocycles. In addition, N-glucuronidation rates in humans are reported to be much higher than in animals, largely due to UGT1A4 and additionally UGT2B10 [2]. This species difference in UGT activities can therefore be a barrier for accurate prediction of in vivo glucuronidation of drugs in humans. In fact, rodents lack a human UGT1A4 homologue gene [3]. Subsequent synthesis of the major metabolite of the LEO compound allowed further investigations to be conducted showing excretion of the N-glucuronide in the bile, its hydrolysis and the subsequent reabsorption of parent. However, this was not predicted to lead to significant enterohepatic recirculation [4].

Figure 1: Glucuronidation of a pyrazole moiety in LEO compound 1 by UGT1A4 to a major metabolite observed at 14% in human hepatocytes in the MetID study.
Further complexity arises from involvement of UGT1A4 as the gene encoding this enzyme is highly polymorphic. Although more than 100 polymorphisms have been identified, the two most commonly studied can result in altered enzyme activity, with UGT1A4*3 increasing glucuronidation and UGT1A4*2 decreasing glucuronidation [5]. For example, the UGT1A4*3 variant has been shown to affect the metabolism of olanzapine, an antipsychotic drug metabolised to four oxidised metabolites and two N-glucuronides. The major metabolite of olanzapine in humans is a tertiary N-glucuronide in which the glucuronic acid moiety is attached to the nitrogen at position 10 of the benzodiazepine ring. Its formation is catalysed by UGT1A4. A quaternary N-linked 4′-N-glucuronide is additionally observed in urine [6]. Studies showed that the N-10 glucuronide was only detected in the plasma and urine of humans and not in mice, rats, or monkeys, with only a trace detected in dog urine [7]. It is resistant to enzymatic and base hydrolysis but was cleaved under acidic conditions.

Figure 2: N-glucuronides of olanzapine. Olanzapine is also subject to phase I metabolism including N-demethylation by CYP1A2, 2-hydroxylation by CYP2D6 and N-oxidation by FMO3.
Carriers of the UGT1A4*3 variant can experience reduced therapeutic efficacy due to ultrarapid glucuronidation, thus higher doses of some UGT1A4 substrates may be required for people carrying the UGT1A4*3 variant, such as seen during posaconazole treatment as an antifungal prophylaxis during treatment of hematological malignancies. [8].
A recent paper describing the metabolism of pexidartinib by CYP3A4 and UGT1A4 revealed that UGT1A4-mediated glucuronidation resulted in generation of an N-glucuronide at a 10% higher exposure than the parent drug [11]. Despite the benign inactive N-glucuronide being the major metabolite, pexidartinib carries a box warning due to drug-induced liver injury (DILI). This is however attributed to reactive metabolites mediated by CYP3A4, resulting in the detection of GSH adducts in HLMs [12]. As in this example, most N-glucuronides of small molecule drugs are innocuous and possess no pharmacological activity. However, individuals possessing loss of function polymorphisms in UGTs responsible for formation of major glucuronides may be subject to reduced clearance and possible flux through alternative routes than result in metabolites of greater concern.

Figure 3: Pexidartinib is metabolised via oxidation, epoxidation and dealkylation by CYP 3A4/5, and glucuronidation by UGT1A4. UGT1A4 catalyses formation of a major inactive N-glucuronide, which is approximately at a 10% higher exposure than pexidartinib after a single dose.
Stability of N-glucuronides
Enterohepatic recycling increases the half-life and the residence time for a drug being recycled, thus increasing systemic exposure and a delay in clearance. Both O– and N-glucuronides can be cleaved back to the active parent compound in the intestines through the action of β-glucuronidases produced by gut bacteria. However not all N-glucuronides are similarly susceptible; and their hydrolysis may also depend on the source of the β-glucuronidase enzyme [9].
The formation of unstable N-glucuronides of aromatic amine containing drugs was previously linked to bladder cancer after hydrolysis back to the parent drug under the acidic conditions in the bladder. Conversely, and interestingly, UGTs present in the bladder can protect against accumulation of toxins since they are highly expressed on the surface of bladder tissue, particularly in females where it is postulated that the lower incidence of bladder cancer is related to increased UGT isoform activity. UGT1A enzymes were shown to be down-regulated in several cancers, and mutations in UGT genes enhance the risk of bladder cancer in smokers [10].
A route to be avoided or a desired route of metabolism?
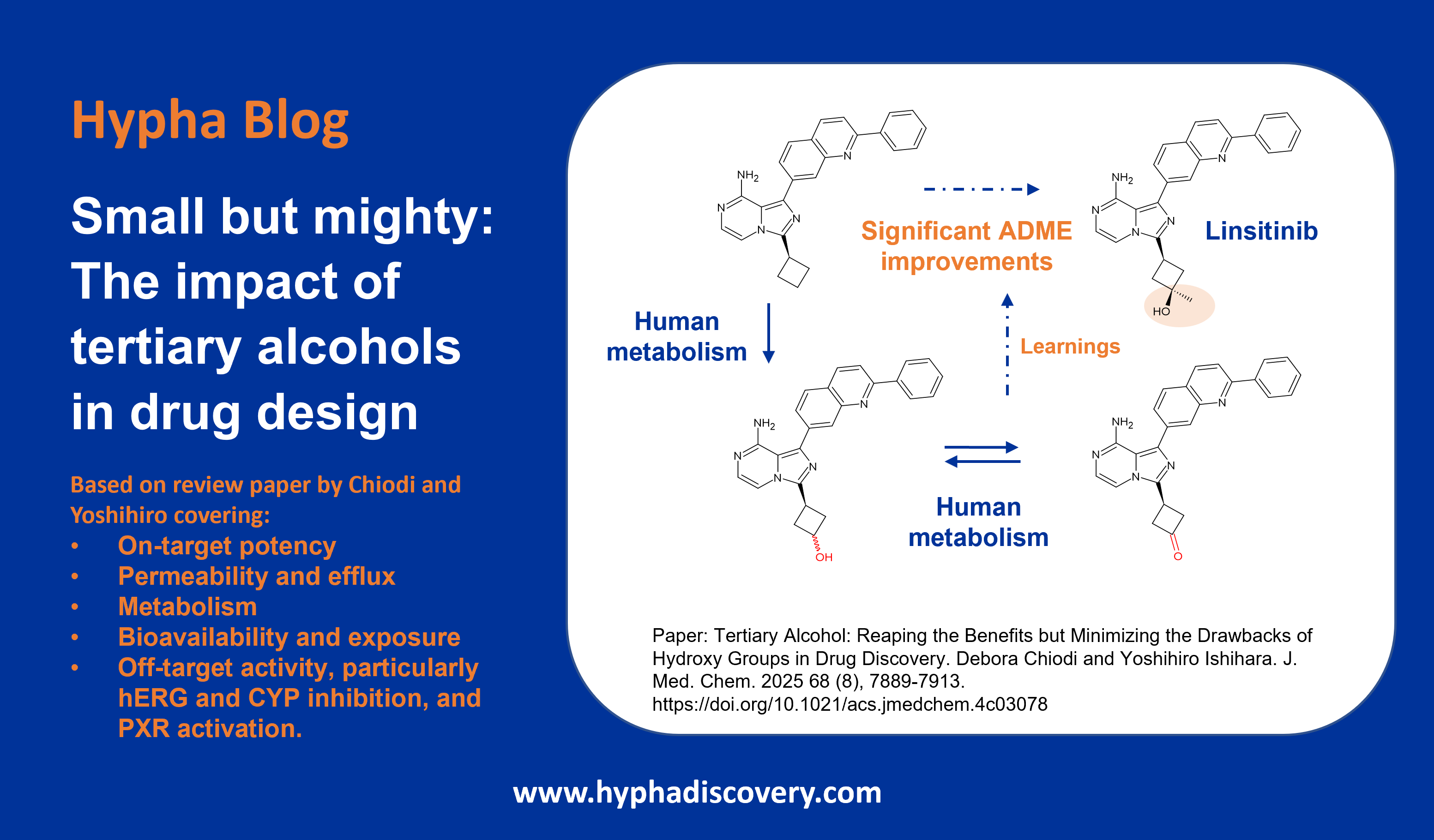
We have worked on some projects where (non-acyl) glucuronidation was desired as a means to a benign route of metabolism, balanced with an appropriate clearance, since most O– and N-glucuronides do not result in reactive metabolites and are readily excreted. This is in contrast to the view taken by some chemists that moieties deemed vulnerable to glucuronidation are to be avoided. Where excessive glucuronidation has been observed, structural modifications have been incorporated to reduce this, such as in the design of a novel class of oxetane indole-amine 2,3-dioxygenase (IDO1) inhibitors [13] Here introduction of a hydroxyl group to improve polarity also had a positive effect on the off-target profile, but which was subject to O-glucuronidation. To curb glucuronidation at that position a gem-dimethyl group was incorporated which not only boosted metabolic stability, but also resulted in increased potency of the final lead compound.

Figure 4: Optimisation of an IDO1 inhibitor incorporating installation of a gem-dimethyl group to control glucuronidation of a key hydroxyl group.
References
[1] Metabolite Transport in DDIs, Toxicity, and Efficacy. Maciej J. Zamek-Gliszczynski, Xiaoyan Chu, Joseph W. Polli, Mary F. Paine and Aleksandra Galetin (2014). Drug Metabolism and Disposition 42 (4) 650-664; https://doi.org/10.1124/dmd.113.055558.
[2] N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Sanna Kaivosaari, Moshe Finel & Mikko Koskinen (2011). Xenobiotica, 41:8, 652-669; https://doi.org/10.3109/00498254.2011.563327.
[3] Glucuronidation of drugs in humanized UDP-glucuronosyltransferase 1 mice: Similarity with glucuronidation in human liver microsomes. Yuki Kutsuno, Kyohei Sumida, Tomoo Itoh, Robert H. Tukey, Ryoichi Fujiwara. Pharma Res Per 1 (1), 2013, e00002; https://doi.org/10.1002/prp2.2
[4] Discovery of an oral, rule-of-5 compliant, IL-17A protein-protein interaction modulator (PPIm) for the treatment of psoriasis and other inflammatory diseases. Mark Andrews, presentation at the 3rd RSC Anglo-Nordic Medicinal Chemistry Symposium, 13th-16th June 2023.
[5] https://genesight.com/white-papers/get-to-know-a-gene-ugt1a4/
[6] Disposition and biotransformation of the antipsychotic agent olanzapine in humans. Kassahun K, Mattiuz E, Nyhart E Jr, Obermeyer B, Gillespie T, Murphy A, et al. (1997). Drug Metab Dispos. 25(1):81-93. PMID: 9010634.
[7] Olanzapine 10-N-glucuronide. A tertiary N-glucuronide unique to humans. Kassahun K, Mattiuz E, Franklin R, Gillespie T. (1998). Drug Metab Dispos. 26(9):848-55. PMID: 9733662.
[8] The Genetic Polymorphism UGT1A4*3 Is Associated with Low Posaconazole Plasma Concentrations in Hematological Malignancy Patients Receiving the Oral Suspension. Suh HJ, Yoon SH, Yu KS, Cho JY, Park SI, Lee E et al. (2018). Antimicrob Agents Chemother. 26;62(7):e02230-17; https://doi.org/10.1128/aac.02230-17
[9] Stability and enzymatic hydrolysis of quaternary ammonium-linked glucuronide metabolites of drugs with an aliphatic tertiary amine-implications for analysis. Kowalczyk I, Hawes EM, McKay G. (2000). J Pharm Biomed Anal. 22(5):803-11; https://doi.org/10.1016/S0731-7085(00)00244-2
[10] Glucuronidation and UGT isozymes in bladder: new targets for the treatment of uroepithelial carcinomas? Sundararaghavan VL, Sindhwani P, Hinds TD Jr. (2017). Oncotarget. 8(2):3640-3648; https://doi.org/10.18632/oncotarget.12277
[11] Pharmacokinetics of the Multi-kinase Inhibitor Pexidartinib: Mass Balance and Dose Proportionality. Zahir, H., Greenberg, J., Hsu, C., Watanabe, K., Makino, C., He, L. and LaCreta, F. (2023). Clin Pharmacol Drug Dev, 12: 159-167. https://doi.org/10.1002/cpdd.1186
[12] Identifying the Reactive Metabolites of Tyrosine Kinase Inhibitor Pexidartinib In Vitro Using LC–MS-Based Metabolomic Approaches. Xuan Qin, Yong Wang, Kevin R. MacKenzie, John M. Hakenjos, Si Chen, Saleh M. Khalil et al.(2023). Chemical Research in Toxicology 36 (8), 1427-1438; https://doi.org/10.1021/acs.chemrestox.3c00164
[13] Oxetane Promise Delivered: Discovery of Long-Acting IDO1 Inhibitors Suitable for Q3W Oral or Parenteral Dosing. Derun Li, David L. Sloman, Abdelghani Achab, Hua Zhou, Meredeth A. McGowan, Catherine White et al. (2022). Journal of Medicinal Chemistry 65 (8), 6001-6016; https://doi.org/10.1021/acs.jmedchem.1c01670