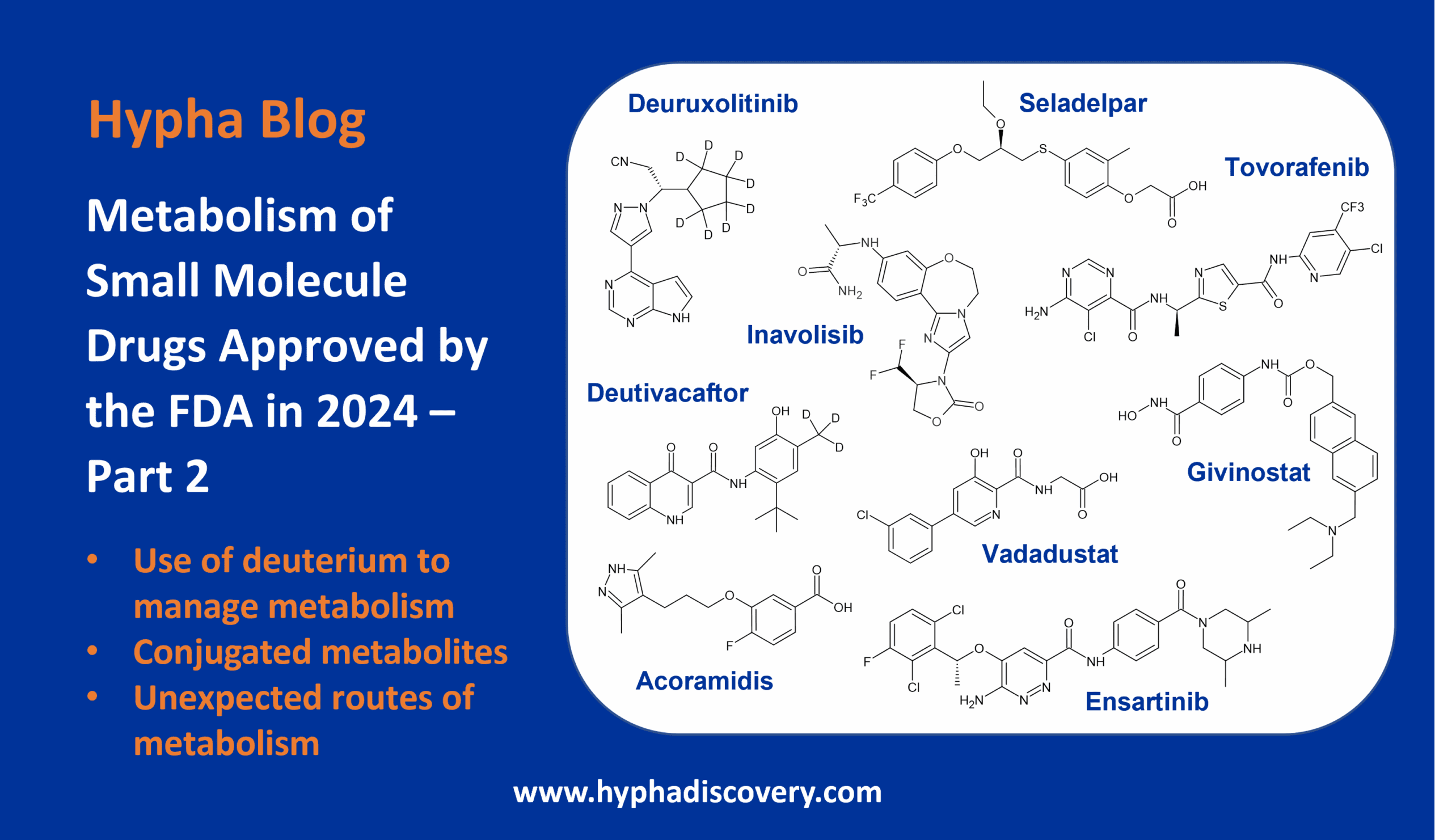
Metabolism of de novo-designed macrocyclic drugs approved by the FDA
By Julia Shanu-Wilson
To date, only 4% (67) of FDA approved drugs are macrocycles [1]. However, in the quest to tackle challenging disease targets with difficult drug binding sites, there is revived interest in developing this compound type to offer unique solutions.
Current macrocycles in clinical use generally focus on treatment of infectious diseases, cancer and auto-immune disorders. Previously natural products have provided the source of up to 90% of macrocyclic drugs, not surprising given that 20% of natural product derived ring systems are macrocyclic [2]. However, more de novo designed molecules in this “beyond the Rule of 5 chemical space” have emerged, with the first in this class, plerixafor, approved in oncology in 2008. Still, of the 34 macrocycles currently in clinical trials, only 18% are de novo designed.
Macrocycles contain 12 or more atoms in a ring system, which provides conformational restraint useful for selective and potent binding, particularly for large, flat, featureless, difficult-to-drug binding sites. This was illustrated nicely by optimized macrocyclic Mcl-1 Inhibitors, which are highly preorganized in solution and display exquisite potency and selectivity as PPI inhibitors, suggesting that ligand rigidity achieved by macrocyclization is an important component of their activity [3]. Not only this, the reduction in rotatable bonds and consequent reduction in susceptibility to metabolism is an additional benefit [4].
With metabolic stability in mind, we take a look at the metabolism of some FDA approved synthetic macrocyclic drugs, and a few of those that were/are in clinical development where metabolism data was available.
A multitude of macrocyclic antiviral drugs
The antiviral drug simeprevir (TMC435) was approved for the treatment of chronic hepatitis C in 2013 but has since been discontinued in favour of more effective drugs. It’s metabolised to 3 main metabolites by CYP3A with potential additional involvement of CYP2C8 and CYP2C19.
Although in vitro studies showed low metabolic activity, more than 20 metabolites could be identified, resulting from two main routes. O-demethylation was more prevalent in animals with oxidation the preferred route in monkey and man, however no major human disproportionate metabolites were seen in vivo. The major metabolites reported in vivo in plasma from animals and human were M18 (O-demethylation at the aromatic position) and M21 (oxidation at the macrocycle moiety) [5].
The human radiolabeled ADME study revealed that M21 was the most abundant circulating metabolite in plasma (7.96% of mean plasma AUC0-24h of the parent) [6]. Since M21 represented less than 10% of unchanged drug, systemic exposure to M21 was not assessed in the safety evaluation studies, and did not appear to accumulate in man.

Figure 1 Metabolism of simeprevir
Glecaprevir (ABT493/A-1282576) is another antiviral drug for the treatment of chronic hepatitis C which also undergoes limited CYP3A4/5 mediated metabolism in vitro, and for which all human metabolites were present in at least one animal species.
A combination therapy of glecaprevir and pibrentasvir has recently been implicated in a case of liver injury. It was hypothesized that as glecaprevir is largely metabolized by the liver, the production of toxic or immunogenic metabolites was a possible source of injury [7]. Interestingly, minor (<10% of total drug related materials) metabolites present in human plasma at steady state including one defluorinated and eight oxidised metabolites. It is not clear what the cumulative amount of different metabolite related species would be. Cys and GSH conjugates were additionally observed in dog. It should be noted that the patient was also on pantoprazole and budesonide, which are also metabolized by the liver [8].

Figure 2 Human metabolism of glecaprevir
Involvement of gut bacteria
Grazoprevir (MK5172) is also used as part of combination therapy to treat chronic hepatitis C. Another metabolically stable macrocycle, no circulating metabolites were detected in human plasma. Again, CYP3A is responsible for elimination with 5 metabolites resulting from hydroxylation, one from O-dealkylation and one from oxidative loss of vinylcyclopropylsulfonamide. Additionally, a GSH conjugate was identified in-vitro and in-vivo arising from CYP3A4 catalyzed bioactivation, but proposed as only a minor pathway in humans [9].
Interestingly three metabolites (M10, M11a and M11b) that were not observed in vitro were identified in human faeces, the likely result of gut bacterial metabolism (Figure 3). Two of these (M11a and M11b) were only observed in humans but thought not to be absorbed in the GI tract. In addition, a human specific oxidative metabolite (M14) was identified in human feces comprising ~4% of total grazoprevir-related material. The presence of this minor human specific metabolite was not thought to pose a significant clinical safety risk.

Figure 3 Human specific metabolites of grazoprevir in faeces
Another antiviral drug where gut metabolism is involved in clearance is paritaprevir. Paritaprevir is part of a three-combination agent and is administered with a low dose of ritonavir to achieve drug concentrations suitable for once-daily dosing for the treatment of patients infected with hepatitis C genotype 1.
Similar to the other macrocycle antivirals highlighted, paritaprevir is metabolised predominantly by CYP3A4 and to a lesser extent by CYP3A5, with the parent drug the major component in plasma. At least 5 minor metabolites (M2, M29, M3, M13, and M6) of paritaprevir have been identified in circulation that accounted for approximately 10% of plasma radioactivity in the human radiolabelled study [10].
The biotransformation of paritaprevir in humans involves two main routes; CYP mediated oxidation on the olefinic linker, the phenanthridine group, the methylpyrazinyl group (or combinations thereof), and amide hydrolysis at the acyl cyclopropane-sulfonamide moiety and the pyrazine-2-carboxamide moiety. Paritaprevir was primarily eliminated through the biliary-fecal route followed by microflora-mediated sulfonamide hydrolysis to M29 as a major component in feces (approximately 60% of dose).

Figure 4 Biotransformation of paritaprevir in humans
The low clearance hepatitis C antiviral voxilaprevir (GS-9857) is slowly turned over by CYP3A and is moderately metabolized with approximately 40% of the dose recovered in feces as parent. Major metabolites in the faeces include those derived from oxidation (M21, 5.4% and M25, 3.8% of dose administered) hydrolysis (M19, 22.1%) and dehydrogenation (M9, 7.5%) biotransformation pathways [11]. There were no human specific metabolites.
Rounding off this set of antivirals is danoprevir (ITMN-191 / RG7227), a macrocyclic peptidomimetic inhibitor of NS3/4A HCV protease. In contrast to some of the other antivirals already described, danoprevir is cleared by several CYPs, including CYP3A4 and CYP2C19 [12].
Synthetic macrocycles in oncology
Pactritinib is a multi-kinase JAK2/FLT3 inhibitor used for the treatment of primary and secondary myelofibrosis, approved by the FDA in 2022. It’s extensively biotransformed to 12 metabolites through multiple mechanisms; oxidation, N-dealkylation, O-dealkylation, hydrolysis, and dehydrogenation. CYP3A4 is involved in clearance to two major metabolites M1 and M2, representing 9.6% and 10.5% of parent drug exposure respectively. M1 results from oxidation of the pyrrole ring and M2 from O-dealkylation of the side chain.
Other identified metabolites include M3, resulting from a different oxidation in the pyrrole ring system, and M4 resulting from reduction of the double bond in the macrocyclic ring system. In human faeces M2 was the predominant metabolite (~24% of dose), whilst glucuronidation was observed to M7, the major metabolite in urine (~3% of dose).

Figure 5 Identified CYP3A4 metabolites of pacritinib in human plasma
Oxidative cleavage and N-glucuronidation
Whilst many macrocyclic drugs appear to be metabolised only by CYP3A4, lorlatinib is additionally metabolised by UGT1A4 with only minor contributions from other metabolizing enzymes (CYP2C8, CYP2C19, CYP3A5, and UGT1A3). Lorlatinib is an interesting third-generation inhibitor of anaplastic lymphoma kinase (ALK), with which higher exposures in the CNS are achieved compared to previous generations of inhibitors. A structure-based design process led to improved CNS penetration as a consequence of a transition from the acyclic crizotinib predecessor to a macrocycle. The resulting macrocyclic structure was found to be associated with improved metabolic stability and low propensity for P-gp efflux compared to the acyclic analog [13].
The human [14C] mass balance study confirmed the main biotransformation pathways of oxidation (N-demethylation, N-oxidation) and N-glucuronidation, but surprisingly uncovered an unanticipated, intramolecular oxidative cleavage pathway of lorlatinib, yielding a major human circulating benzoic acid metabolite (M8), and an unlabeled pyrido-pyrazole substructure. The fate of unknown metabolites associated with cleavage of the amide and aromatic ether bonds of lorlatinib necessitated a second ADME study where the radiolabel was positioned on the pyrazole ring [14, 15]. M8 accounted for 21% of the circulating radioactivity in plasma, but was found to be pharmacologically inactive.

Figure 6 Biotransformation of lorlatinib in humans
Macrocycles in the clinic

Acyl glucuronide of AZD5991
AZD5991 is a macrocyclic selective inhibitor of Mcl-1 that was previously in development for treatment of hematological malignancies. Several metabolites were observed with an S-oxide and an acyl glucuronide being major human metabolites. LC-MS data indicated that the acyl glucuronide did not undergo acyl migration under physiological conditions [16].
Metabolism data on other macrocycles in clinical trials is not yet publicly available but where information is available the drugs appear to be minimally metabolised, such as odalasvir an investigational new drug in development for the treatment of hepatitis C, or via CYP3A4 such as milvexian, an oral factor XIa inhibitor for the prevention and treatment of major thrombotic conditions. Zotiraciclib, an oral multi-cyclin dependent kinase inhibitor for the treatment of brain cancer, is additionally metabolised by CYP1A2 as well as CYP3A4 [17]. We await to see what other biotransformation data emerges on synthetic macrocycles as more information is publicly disclosed as they advance through development.
References
[1] Macrocycles in Drug Discovery─Learning from the Past for the Future. Garcia Jimenez et al., 2023. Journal of Medicinal Chemistry, 66 (8), 5377-5396. https://doi.org/10.1021/acs.jmedchem.3c00134
[2] Ring systems in natural products: structural diversity, physicochemical properties, and coverage by synthetic compounds. Chen et al., 2022. Nat. Prod. Rep., 39, 1544-1556. https://doi.org/10.1039/D2NP00001F
[3] Free Ligand 1D NMR Conformational Signatures To Enhance Structure Based Drug Design of a Mcl-1 Inhibitor (AZD5991) and Other Synthetic Macrocycles. Balazs et al., 2019. Journal of Medicinal Chemistry 62 (21), 9418-9437. https://doi.org/10.1021/acs.jmedchem.9b00716
[4] Macrocyclization via Ring-Closing Metathesis in Drug Discovery & Process Chemistry. Drug Hunter article, June 2023. https://drughunter.com/macrocyclization-via-ring-closing-metathesis-in-drug-discovery-process-chemistry/
[5] European Medicines Agency (EMA), Committee for Medicinal Products for Human Use (CHMP), European Public Assessment Report (EPAR): Olysio (Simeprevir); p.20 (March 20, 2014). https://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002777/WC500167870.pdf
[6] PHARMACOLOGY/TOXICOLOGY NDA REVIEW AND EVALUATION Study NC219 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/205123Orig1s000PharmR.pdf
[7] Acute Liver Injury due to Glecaprevir/Pibrentasvir in a Patient with Chronic Hepatitis C Virus Infection without Cirrhosis. Ayushi Jain and Khalid Mumtaz, 2022. Avicenna J Med 12:154–156. https://www.thieme-connect.com/products/ejournals/html/10.1055/s-0042-1750716
[8] Glecaprevir/pibrentasvir-associated acute liver injury in non-cirrhotic, chronic HCV infection without HBV co-infection. Hammami et al., 2019. BMJ Case Reports CP 2019;12:e226622. http://dx.doi.org/10.1136/bcr-2018-226622
[9] PHARMACOLOGY/TOXICOLOGY NDA REVIEW AND EVALUATION. APPLICATION NUMBER: 208261Orig1s000 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208261Orig1s000PharmR.pdf
[10] SUMMARY OF PRODUCT CHARACTERISTICS for Viekirax https://www.ema.europa.eu/en/documents/product-information/viekirax-epar-product-information_en.pdf
[11] CLINICAL PHARMACOLOGY AND BIOPHARMACEUTICS REVIEW(S). APPLICATION NUMBER: 209195Orig1s000 https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209195Orig1s000ClinPharmR.pdf
[12] Preclinical Characteristics of ITMN-191, an Orally Active Inhibitor of the HCV NS3/4A Protease Nominated for Preclinical Development. 2006. https://www.natap.org/2006/DDW/DDW_30.htm
[13] A user’s guide to lorlatinib. Nagasaka et al., 2020. Critical Reviews in Oncology/Hematology, 151, 102969, https://doi.org/10.1016/j.critrevonc.2020.102969.
[16] Structural Identification and Synthesis of the Acyl Glucuronide of the Macrolide Mcl-1 Inhibitor AZD5991. Gangl et al., 2023. Poster at https://hyphadiscovery.wpengine.com/poster/structural-identification-and-synthesis-of-the-acyl-glucuronide-of-the-macrolide-mcl-1-inhibitor-azd5991/
[17] Phase I Study of Zotiraciclib in Combination with Temozolomide for Patients with Recurrent High-grade Astrocytomas. Wu et al., 2021. Clin Cancer Res 27 (12): 3298–3306. https://doi.org/10.1158/1078-0432.CCR-20-4730